From Wikipedia, the free encyclopedia
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities.[1] The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are now often referred to as congenital metabolic diseases or inherited metabolic disorders.[2] To this concept it’s possible to include the new term of Enzymopathy. This term was created following the study of Biodynamic Enzymology, a science based on the study of the enzymes and their derivated products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the «one gene-one enzyme» hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.[3]
Classification and symptoms of metabolic diseases[edit]
Traditionally the inherited metabolic diseases were classified as disorders of carbohydrate metabolism, amino acid metabolism, organic acid metabolism, or lysosomal storage diseases.[4] In recent decades, hundreds of new inherited disorders of metabolism have been discovered and the categories have proliferated. Following are some of the major classes of congenital metabolic diseases, with prominent examples of each class.[5]
- Disorders of carbohydrate metabolism
- glycogen storage disease
- G6PD deficiency
- Disorders of amino acid metabolism
- phenylketonuria
- maple syrup urine disease
- glutaric acidemia type 1
- Urea Cycle Disorder or Urea Cycle Defects
- Carbamoyl phosphate synthetase I deficiency
- Disorders of organic acid metabolism (organic acidurias)
- alkaptonuria
- Combined malonic and methylmalonic aciduria (CMAMMA)
- 2-hydroxyglutaric acidurias
- Disorders of fatty acid oxidation and mitochondrial metabolism
- Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCADD)
- Disorders of porphyrin metabolism
- acute intermittent porphyria
- Disorders of purine or pyrimidine metabolism
- Lesch–Nyhan syndrome
- AMPD1 Deficiency (MADD)
- Disorders of steroid metabolism
- lipoid congenital adrenal hyperplasia
- congenital adrenal hyperplasia
- Disorders of mitochondrial function
- Kearns–Sayre syndrome
- Disorders of peroxisomal function
- Zellweger syndrome
- Lysosomal storage disorders
- Gaucher’s disease
- Niemann–Pick disease
Because of the enormous number of these diseases the wide range of systems affected badly, nearly every «presenting complaint» to a healthcare provider may have a congenital metabolic disease as a possible cause, especially in childhood and adolescence. The following are examples of potential manifestations affecting each of the major organ systems.
- Growth failure, failure to grow, loss of weight
- Ambiguous genitalia, delayed puberty, precocious puberty
- Developmental delay, seizures, dementia, encephalopathy, stroke
- Deafness, blindness, pain agnosia
- Skin rash, abnormal pigmentation, lacking of pigmentation, excessive hair growth, lumps and bumps
- Dental abnormalities
- Immunodeficiency, low platelet count, low red blood cell count, enlarged spleen, enlarged lymph nodes
- Many forms of cancer
- Recurrent vomiting, diarrhea, abdominal pain
- Excessive urination, kidney failure, dehydration, edema
- Low blood pressure, heart failure, enlarged heart, hypertension, myocardial infarction
- Liver enlargement, jaundice, liver failure
- Unusual facial features, congenital malformations
- Excessive breathing (hyperventilation), respiratory failure
- Abnormal behavior, depression, psychosis
- Joint pain, muscle weakness, cramps
- Hypothyroidism, adrenal insufficiency, hypogonadism, diabetes mellitus
Diagnostic[edit]
Dozens of congenital metabolic diseases are now detectable by newborn screening tests, especially expanded testing using mass spectrometry.[6] Gas chromatography–mass spectrometry-based technology with an integrated analytics system has now made it possible to test a newborn for over 100 mm genetic metabolic disorders. Because of the multiplicity of conditions, many different diagnostic tests are used for screening. An abnormal result is often followed by a subsequent «definitive test» to confirm the suspected diagnosis.

Common screening tests used in the last sixty years:
- Ferric chloride test (detects abnormal metabolites in urine)
- Ninhydrin paper chromatography (detects abnormal amino acid patterns)
- Guthrie test (detects excessive amounts of specific amino acids in blood) The dried blood spot can be used for multianalyte testing using Tandem Mass Spectrometry (MS/MS). This given an indication for a disorder. The same has to be further confirmed by enzyme assays, IEX-Ninhydrin, GC/MS or DNA Testing.
- Quantitative measurement of amino acids in plasma and urine
- IEX-Ninhydrin post-column derivitization liquid ion chromatography (detects abnormal amino acid patterns and quantitative analysis)
- Urine organic acid analysis by gas chromatography–mass spectrometry
- Plasma acylcarnitine analysis by mass spectrometry
- Urine purine and pyrimidine analysis by gas chromatography-mass spectrometry
Specific diagnostic tests (or focused screening for a small set of disorders):
- Tissue biopsy: liver, muscle, brain, bone marrow
- Skin biopsy and fibroblast cultivation for specific enzyme testing
- Specific DNA testing
A 2015 review reported that even with all these diagnostic tests, there are cases when «biochemical testing, gene sequencing, and enzymatic testing can neither confirm nor rule out an IEM, resulting in the need to rely on the patient’s clinical course».[7] A 2021 review showed that several neurometabolic disorders converge on common neurochemical mechanisms that interfere with biological mechanisms also considered central in ADHD pathophysiology and treatment. This highlights the importance of close collaboration between health services to avoid clinical overshadowing.[8]
Treatment[edit]
In the middle of the 20th century the principal treatment for some of the amino acid disorders was restriction of dietary protein and all other care was simply management of complications. In the past twenty years, new medications, enzyme replacement, gene therapy, and organ transplantation have become available and beneficial for many previously untreatable disorders. Some of the more common or promising therapies are listed:
- Dietary restriction
- E.g., reduction of dietary protein remains a mainstay of treatment for phenylketonuria and other amino acid disorders
- Dietary supplementation or replacement
- E.g., oral ingestion of cornstarch several times a day helps prevent people with glycogen storage diseases from becoming seriously hypoglycemic.
- Medications
- E.g., Nitisisone prevents the formation of toxic metabolites for patients with Tyrosinemia Type I and enables normal growth and development in combination with a low-protein diet
- Vitamins
- E.g., thiamine supplementation benefits several types of disorders that cause lactic acidosis.
- Intermediary metabolites, compounds, or drugs that facilitate or retard specific metabolic pathways
- Dialysis[disambiguation needed]
- Enzyme replacement E.g. Acid-alpha glucosidase for Pompe disease
- Gene therapy
- Bone marrow or organ transplantation
- Treatment of symptoms and complications
- Prenatal diagnosis
Epidemiology[edit]
In a study in British Columbia, the overall incidence of the inborn errors of metabolism were estimated to be 40 per 100,000 live births or 1 in 2,500 births,[9] overall representing more than approximately 15% of single gene disorders in the population.[9] While a Mexican study established an overall incidence of 3.4: 1000 live newborns and a carrier detection of 6.8:1000 NBS.[10]
| Type of inborn error | Incidence | |
|---|---|---|
| Disease involving amino acids (e.g. PKU, Tyrosinemia), organic acids, primary lactic acidosis, galactosemia, or a urea cycle disease |
24 per 100 000 births[9] | 1 in 4,200[9] |
| Lysosomal storage disease | 8 per 100 000 births[9] | 1 in 12,500[9] |
| Peroxisomal disorder | ~3 to 4 per 100 000 of births[9] | ~1 in 30,000[9] |
| Respiratory chain-based mitochondrial disease | ~3 per 100 000 births[9] | 1 in 33,000[9] |
| Glycogen storage disease | 2.3 per 100 000 births[9] | 1 in 43,000[9] |
References[edit]
- ^ MedlinePlus Encyclopedia: Inborn errors of metabolism
- ^ «Inherited metabolic disorders — Symptoms and causes». Mayo Clinic.
- ^ Garrod, Archibald E (1923). Inborn errors of metabolism. OCLC 1159473729.[page needed][non-primary source needed]
- ^ Bartolozzi, Giorgio (2008). «Errori congeniti del metabolismo» [Inborn errors of metabolism] (PDF). Pediatria: principi e Pratica clinica [Pediatrics: Principles and Clinical Practice] (in Italian). pp. 361–386. ISBN 978-88-214-3204-0. OCLC 884592549.
- ^ Sghirlanzoni, Angelo (2010). Terapia delle malattie neurologiche. doi:10.1007/978-88-470-1120-5. ISBN 978-88-470-1119-9.
- ^ Geerdink, R.B; Niessen, W.M.A; Brinkman, U.A.Th (March 2001). «Mass spectrometric confirmation criterion for product-ion spectra generated in flow-injection analysis». Journal of Chromatography A. 910 (2): 291–300. doi:10.1016/s0021-9673(00)01221-8. PMID 11261724.
- ^ Vernon, Hilary J. (1 August 2015). «Inborn Errors of Metabolism: Advances in Diagnosis and Therapy». JAMA Pediatrics. 169 (8): 778–782. doi:10.1001/jamapediatrics.2015.0754. PMID 26075348.
- ^ Cannon Homaei S, Barone H, Kleppe R, Betari N, Reif A, Haavik J (2021). «ADHD symptoms in neurometabolic diseases: Underlying mechanisms and clinical implications». Neuroscience and Biobehavioral Reviews. 132: 838–856. doi:10.1016/j.neubiorev.2021.11.012. PMID 34774900. S2CID 243983688.
- ^ a b c d e f g h i j k l Applegarth, Derek A.; Toone, Jennifer R.; Lowry, R. Brian (1 January 2000). «Incidence of Inborn Errors of Metabolism in British Columbia, 1969–1996». Pediatrics. 105 (1): e10. doi:10.1542/peds.105.1.e10. PMID 10617747.
- ^ Navarrete-Martínez, Juana Inés; Limón-Rojas, Ana Elena; Gaytán-García, Maria de Jesús; Reyna-Figueroa, Jesús; Wakida-Kusunoki, Guillermo; Delgado-Calvillo, Ma. del Rocío; Cantú-Reyna, Consuelo; Cruz-Camino, Héctor; Cervantes-Barragán, David Eduardo (May 2017). «Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three-year findings from a screening program in a closed Mexican health system». Molecular Genetics and Metabolism. 121 (1): 16–21. doi:10.1016/j.ymgme.2017.03.001. PMID 28302345.
Further reading[edit]
- Price, Nicholas C; Stevens, Lewis (1996). Principi di enzimologia [Principles of enzymology] (in Italian). A. Delfino. ISBN 978-88-7287-100-3. OCLC 879866185.
- Mazzucato, Fernando; Giovagnoni, Andrea (2019). Manuale di tecnica, metodologia e anatomia radiografica tradizionali [Manual of traditional radiographic technique, methodology and anatomy] (in Italian). Piccin. ISBN 978-88-299-2959-7. OCLC 1141547603.
- Torricelli, P; Antonelli, F; Ferorelli, P; Borromeo, I; Shevchenko, A; Lenzi, S; De Martino, A (March 2020). «Oral nutritional supplement prevents weight loss and reduces side effects in patients in advanced lung cancer chemotherapy». Amino Acids. 52 (3): 445–451. doi:10.1007/s00726-020-02822-7. PMID 32034492. S2CID 211053578.
External links[edit]
- Portal of Chemistry (Italian)
Термин «врожденные ошибки метаболизма» ( “inborn errors of metabolism”) (IEM) был впервые предложен сэром Арчибальдом Гарродом ( Archibald Garrod) в 1908 году. Этим термином автор пытался описать те заболевания, которые вызваны блокированием метаболического пути из-за недостаточной активности конкретного фермента.
Диагноз врожденных нарушений обмена веществ (IEM) играет большую роль в педиатрии. Благодаря внедрению скрининга новорожденных (NBS), диагностика многих IEM стала относительно легкой с использованием лабораторных биомаркеров. Для большинства IEM ранняя диагностика предотвращает появление серьезных клинических симптомов, тем самым снижая заболеваемость и смертность. Однако из-за молекулярной, биохимической и клинической вариабельности IEM не все нарушения, включенные в программы NBS, будут обнаружены и диагностированы только скринингом.
В последние годы прогресс в таких технологиях, как тандемная масс-спектрометрия (МS / МS) и секвенирование следующего поколения (NGS), в которых используется стратегия массивного параллельного секвенирования, значительно расширили наши знания о IEM и метаболических нарушениях в целом. Эти новые технологии также позволили расширить и улучшить скрининг новорожденных (NBS) в глобальном масштабе. Приблизительно 80% расстройств, проверенных с помощью NBS являются IEM.
IEM можно классифицировать на две широкие категории: те, которые влияют на выработку энергии, и те, которые влияют на синтез или распад конкретных молекул или соединений. Хотя углеводы, жиры и белки используются в качестве источников энергии, степень использования определенного топлива зависит от типа органа или ткани. Нарушение метаболического процесса, который влияет на один тип энергии , приводит к увеличению использования альтернативного типа топлива; этот компенсаторный сдвиг в производстве энергии может привести к патологии аномальных метаболитов, наблюдаемой у пациентов с этими нарушениями, что является ключом к постановке диагноза. Благодаря комплексной регуляции обменных процессов во всем организме , накопление субстрата из заблокированной метаболической стадии будет задействовать альтернативные метаболические пути, которые обычно минимально используются в нормальных условиях, что приводит к увеличению производства и накопления потенциально вредных промежуточных метаболитов у пациентов с IEM. Эти промежуточные продукты могут также нарушать нормальный обмен веществ посредством активации или торможения ферментативных процессов или конкурентного действия, что приводит к появлению дополнительных клинических симптомов и патогномоничных паттернов повышенного уровня анализируемых веществ.
Большинство органических ацидурий, аминокислотных патологий, пероксисомных расстройств, лизосомальных нарушений накопления, нарушений накопления гликогена (GSD) и нарушений окисления митохондриальных жирных кислот являются примерами дефектных путей, при которых специфические ферменты расщепляют субстраты (гликоген, органические кислоты, аминокислоты или жирные кислоты) для производства энергии или для генерации основных строительных блоков, используемых в последующих процессах синтеза (например, синтез креатина). Напротив, порфирии, дефицит церебрального креатина и врожденные нарушения гликозилирования являются примерами дефектов в синтетических путях, которые влияют на выработку гема, креатина и гликопротеинов соответственно. Транспортеры и канальные белки, которые мобилизуют субстрат, попадают в обе категории.
Клинические фенотипы IEM являются широкими и часто неспецифичными, имитируя более распространенные состояния, причем, появление симптомов может проявиться в любом возрасте от плода до взрослого человека. Поскольку пациенты с более легкими мутациями и едва различимыми биохимическими фенотипами могут быть пропущены по предельным значениям, используемым для определения положительных результатов скрининга новорожденных , нормальный скрининг новорожденных не должен исключать эти расстройства из дифференциального диагноза у пациента, клиническая картина которого наводит на мысль о наследственном дефекте метаболизма. Клинические и биохимические фенотипы IEM у детей изучались десятилетиями; наоборот, варианты позднего или взрослого начала оставались в значительной степени нераспознанными. В последние годы взрослые формы многих наследственных метаболических нарушений все чаще идентифицируются как истинные легкие фенотипы, где симптомы в детстве не были достаточно серьезными, чтобы заслуживать изучения.
При оценке пациента на предмет возможной IEM обычные лабораторные анализы могут выявить основные паттерны, подозрительные для метаболического дефекта. Общие данные включают гипокетотическую гипогликемию, лактоацидоз, метаболический ацидоз, кетоз, гипераммонемию или метаболический ацидоз в сочетании с гипераммонемией. Оценка этих результатов анализа крови и мочи в сочетании с клинической картиной может сузить акцент на конкретном подмножестве метаболических нарушений. Среди подсказок, которые должны побудить клиницистов заподозрить IEM, есть такие сценарии, как больные новорожденные с ухудшением в анамнезе после неосложненной беременности, эпизодами болезни или колеблющимися симптомами летаргии или другими неврологическими симптомами, вызванными интеркуррентным заболеванием или стрессом, мультисистемным вовлечением, неспособность развиваться, задержка развития, прогрессирующие неврологические признаки или странные неврологические симптомы с или без психологических проблем у пациентов, у которых обычная этиология была исключена ( особенно у взрослых). Хотя биохимические генетические и молекулярно-генетические тесты необходимы для подтверждения диагноза, базовые лабораторные тесты все еще важны и часто дают первые ключи к возможно лежащему в основе IEM.
Первым шагом в выборе подходящего лабораторного исследования для исключения наследственногонарушения метаболизма является определение вероятности возникновения этого состояния из-за дефектов метаболизма малых молекул (таких как аминокислот, органических кислот, пуринов и пиримидинов, цикл мочевины, митохондриальный энергетический метаболизм) или нарушения метаболизма органелл (такие как лизосомы или пероксисомы). Пациенты с низкомолекулярными расстройствами обычно имеют острое начало заболевания, требующее экстренного вмешательства. Базовые лабораторные анализы должны проводиться у каждого ребенка с острым заболеванием, у которого возможно основное нарушение обмена веществ.
Пациенты с нарушениями метаболизма органелл обычно имеют неврологические и нервно-мышечные проявления, органомегалию, дисфункцию печени, с дисморфизмом или без него. Тем не менее, важно иметь в виду, что в некоторых случаях нарушения, влияющие на функцию органелл, могут также сопровождаться метаболическими кризисами, включая гипогликемию и / или метаболический ацидоз, требующие экстренного вмешательства. Кроме того, важно иметь ввиду , что некоторые IEM могут проявляться без метаболического кризиса, угрожающего жизни, причем , с такими необычными симптомами, как образование пузырей на коже после воздействия солнечного света и / или неврологические проявления (например, криптогенная боль в животе, парестезия или психотические эпизоды), как мы это видим в случаях большинства порфирий.
Метаболический ацидоз
Метаболический ацидоз — это нарушение кислотно-щелочного баланса в организме из-за потери бикарбоната, снижения почечной экскреции или увеличения выработки кислот. Определение кислотно-основного состояния важно при оценке состояния пациента с потенциальным наследственным метаболическим дефектом, поскольку высокий метаболический ацидоз с анионным зазором обычно вызывается накоплением органических кислот, включая молочную кислоту, кетоновые тела или необычные кислоты и их производные. Напротив, диарея и ацидоз почечных канальцев являются основными причинами метаболического ацидоза с нормальным разрывом анионов. При наличии ацидоза его следует оценивать в сочетании с другими метаболическими состояниями, такими как гипо- и гипергликемия, кетоз, гиперлактатемия и гипераммонемия.
Большинство IEM, которые представляют в клинической картине метаболический ацидоз и кетоз, являются органическими ацидемиями (то есть, метилмалоновая ацидемия, пропионовая ацидемия, изовалериановая ацидемия). С другой стороны, метаболический ацидоз с гипогликемией и отсутствием кетоза может быть единственной находкой лежащего в основе митохондриального дефекта окисления жирных кислот, где процесс борьбы с гипогликемией нарушен из-за неспособности вырабатывать энергию из метаболизма жирных кислот и увеличения производства -физиологические интермедиаты дикарбоновой кислоты. Метаболический ацидоз и гиперлактатемия в отсутствие повышенных органических кислот, кроме молочной и пировиноградной кислот, могут быть обнаружены при нарушениях метаболизма пировиноградной кислоты, а также при дефектах дыхательной цепи.
Нарушения углеводного обмена
Тяжелая гипогликемия представляет собой опасное для жизни состояние, встречающееся при многих нарушениях обмена веществ, в том числе нарушениях белкового обмена, таких как органическая ацидурия и некоторые аминокислотные патологии. Однако критическая гипогликемия является признаком, обнаруженным при нарушениях, непосредственно влияющих на углеводный обмен, таких как GSD, дефекты глюконеогенеза (дефицит глюкозо-6-фосфатазы, дефицит фруктозо-1,6-бифосфата) и дефекты окисления жирных кислот митохондриями, которые вызывают сильное истощение. циркулирующих и запасных углеводов, вторичных по отношению к дефектному производству альтернативной энергии.
При оценке гипогликемии логический оправданный подход заключается в том, чтобы сначала рассмотреть, является ли пациент «кетотическим» или «некетотическим». Нарушения митохондриального окисления жирных кислот, углеводного обмена, метаболизма кетоновых тел и органических ацидемий могут вызвать гипогликемию. Нарушения митохондриального окисления жирных кислот и кетогенеза, включая дефицит HMG-CoA-лиазы и дефицит HMG-CoA-синтазы, а также гиперинсулинемию, связаны с гипокетотической гипогликемией с или без выраженного метаболического ацидоза, тогда как другие нарушения, такие как органическая ацидемия, нарушение метаболизма тела кетонов и реже болезнь мочи кленового сиропа (MSUD), как правило, вызывает кетотическую гипогликемию.
В нормальных физиологических условиях, когда наступает гипогликемия, происходит одновременное превращение печеночного гликогена в глюкозу и увеличение катаболизма свободных жирных кислот. Дефекты окисления жирных кислот в митохондриях вызывают глубокую гипогликемию из-за истощения запасов глюкозы и гликогена в циркулирующей крови, возникающих из-за неспособности метаболизировать жирные кислоты для удовлетворения потребностей в энергии. При этих дефектах также существует неспособность преодолеть гипогликемию и снижение выработки ацетил-КоА из-за уменьшения потока через спираль бета-окисления, что влияет на выработку кетоновых тел.
При GSD наблюдается нарушение превращения печеночного гликогена в циркулирующую глюкозу во время голодания, что приводит к истощению доступных углеводов; гипогликемия связана с гепатомегалией, дисфункцией печени от легкой до тяжелой степени и гиперлактатемией. Тем не менее, гипогликемия может отсутствовать при GSD типа II (болезнь Помпе или дефицит лизосомной кислоты и мальтазы), поскольку цитоплазматический метаболизм гликогена сохраняется и гликоген накапливается только в лизосомах и на ранних стадиях GSD типа IV.
Гипогликемия также может быть обнаружена при нарушениях углеводного обмена, таких как галактоземия или наследственная непереносимость фруктозы. При классической галактоземии накопленный галактозо-1-фосфат ингибирует фосфоглюкомутазу, нарушая гликолиз, тогда как при наследственной непереносимости фруктозы накопленный фруктозо-1-фосфат ингибирует как глюконеогенез, так и гликогенолиз.
Гипогликемия в постпрандиальном состоянии или после непродолжительного голодания (<4 часа) обычно связана с проблемой чрезмерного использования глюкозы, такой как гиперинсулинизм, тогда как гипогликемия после продолжительного голодания (> 8 часов) наводит на мысль о дефекте окисления жирных кислот. Гипогликемия после голодания средней продолжительности (4–8 часов) может быть вызвана гликогенозом или нарушением, влияющим на глюконеогенез. Другие основные лабораторные результаты также могут быть полезны; например, гипогликемия при наличии фиброза и цирроза печени может быть единственной находкой при наследственной тирозинемии I типа.
Гипераммонемия
Как и гипогликемия, гипераммонемия также угрожает жизни; следовательно, уровень аммиака в плазме должен быть проверен у всех пациентов с изменением сознания и энцефалопатией, особенно, у маленьких детей. Гипераммонемия может быть вызвана многими неметаболическими состояниями, включая заболевание печени, портокавальный шунт, гиперактивность глутаматдегидрогеназы или токсичность вальпроевой кислоты. Однако заметное повышение уровня аммиака, обычно в 10–100 раз превышающее верхний предел нормы, может быть связано с нарушениями цикла мочевины. Хотя некоторые органические ацидемии и нарушения митохондриального окисления жирных кислот также могут вызывать гипераммонемию, она обычно менее значительна.
Аммиак, нейротоксичный побочный продукт дезаминирования аминокислот, превращается в экскретируемую мочевину с помощью цикла мочевины в серии ферментативных стадий, происходящих либо в цитозоле, либо в митохондрии. Хотя цикл мочевины очень эффективен при нормальных условиях, он представляет собой сравнительно хрупкий метаболический процесс, на который могут влиять наследственные метаболические нарушения с помощью различных механизмов.
Нарушения цикла мочевины — это наследственные недостатки любого из ферментов цикла мочевины или продукции аллостерического кофактора N-ацетилглутамина, что приводит к тяжелой первичной гипераммонемии. Гипераммонемия также является относительно распространенной вторичной находкой в органических ацидуриях, где накопленные субстраты или промежуточные органические кислоты ингибируют фермент проксимальный цикл мочевины N-ацетилглутаматсинтазу (NAGS), вызывая общее снижение эффективности детоксикации цикла мочевины. Среди органических ацидурий пропионовая ацидемия и метилмалоновая ацидемия, в частности, могут проявляться перемежающейся вторичной гипераммонемией из-за ингибирующей способности накопленного пропионил-КоА. Метаболические нарушения, при которых циркулирует уровень орнитина, цитруллина, или аргинин снижается из-за почечных потерь или снижение эндогенной продукции также может вызывать гипераммонемию, поскольку все три являются промежуточными циклами мочевины. Когда циркулирующие или внутриклеточные уровни этих аминокислот падают, эффективность цикла мочевины может снижаться, что приводит к гипераммонемии; наиболее глубокие потери аминокислот, важных для цикла мочевины, обнаруживаются при цистинурии и непереносимости лизинурического белка, когда почечная реабсорбция орнитина и аргинина может быть значительно снижена из-за конкуренции за общий переносчик двухосновных аминокислот. Напротив, нарушения окисления митохондриальных жирных кислот могут сопровождаться гипераммонемией вследствие комбинированных эффектов истощения субстрата и ингибирования цикла мочевины токсичными видами ацилкарнитина. Уровень ацетил-КоА, конечного продукта бета-окисления жирных кислот, уменьшается, когда общий поток через путь уменьшается. Ацетил-КоА необходим для производства N-ацетилглутамата, который аллостерически активирует фермент карбамоилфосфат-синтетазу 1 (CPS1); CPS1 преобразует аммиак в карбамоилфосфат на ограничивающей скорость первой стадии цикла мочевины. При некоторых дефектах окисления жирных кислот с длинной цепью ацилирование жирных остатков активного сайта CPS1 непосредственно влияет на способность детоксикации цикла мочевины. Наконец, некоторые расстройства могут вызывать гипераммонемию, вторичную по отношению к повреждению органов. Например, при лизосомных расстройствах, включая мукополисахаридозы, а также при некоторых пероксисомных расстройствах, накопление и хранение сложных крупных молекул в печени приводит к гепатоцеллюлярному повреждению, что, в свою очередь, вызывает снижение эффективности цикла мочевины.
Лактоацидоз
Физиологический баланс циркулирующей молочной кислоты, поддерживаемый продукцией молочной кислоты посредством цитоплазматического гликолиза и многокомпонентного митохондриального потребления, может быть нарушен как неметаболическими, так и метаболическими факторами . Лактоацидоз может возникать из-за увеличения выработки лактата или снижения его метаболизма. У большинства метаболических расстройств с гиперлактатемией наблюдается параллельный кетоз, за исключением дефицита пировиноградной дегидрогеназы, гликогеноза I типа или некоторых нарушений окисления жирных кислот. Лактоацидоз чаще всего вызывается гипоксией тканей из-за плохой циркуляции или недостаточного снабжения кислородом по нескольким причинам, включая кардиогенный или гиповолемический шок. Неметаболические источники гиперлактатемии обычно не сопровождаются кетозом. Однако некоторые IEM, включая органические ацидурии, нарушения метаболизма митохондриальной энергии или дефекты глюконеогенеза, также могут проявляться лактоацидозом.
Как только неметаболическая этиология гиперлактатемии была исключена, наиболее распространенными причинами гиперлактатемии, вторичной по отношению к нарушению митохондриальной энергии токсическими метаболитами, являются нарушения окисления жирных кислот, органические ацидурии и, в очень редких случаях, нарушения цикла мочевины. Другие наследственные причины персистирующей гиперлактатемии включают нарушения метаболизма гликогена, нарушения, влияющие на глюконеогенез, и расстройства, непосредственно влияющие на цикл Кребса или метаболизм пировиноградной кислоты. При дефектах глюконеогенеза, таких как дефицит фруктозы-1, 6-фосфатазы и GSD типа IA [дефицит глюкозо-6-фосфатазы (G-6PD)], пики гиперлактатемии отмечаются при голодании или в гипогликемических ситуациях. . При нарушениях, влияющих на деградацию гликогена, гиперлактатемия достигает пика после приема пищи. Гиперлактатемия может различаться при нарушениях, непосредственно влияющих на метаболизм пировиноградной кислоты. При дефиците пируватдегидрогеназы, дефиците альфа-кетоглутаратдегидрогеназы и нарушениях дыхательной цепи гиперлактатемия обычно возникает в состоянии сытости, тогда как при дефиците пируваткарбоксилазы гиперлактатемия возникает как в состоянии натощак, так и в состоянии сытости. При оценке пациента на предмет гиперлактатемии часто упускают из виду соотношение между молочная кислота / пировиноградная кислота и 3-ОН масляная кислота / ацетоуксусная кислота. Молярное соотношение молочной кислоты / пировиноградной кислоты в плазме коррелирует с отношением NAD + / NADH и может косвенно отражать цитозольное окислительно-восстановительное состояние, тогда как соотношение 3-гидроксибутирная кислота / ацетоуксусная кислота отражает внутримитохондриальное окислительно-восстановительное состояние.
Кетонурия
Кетоновые тела 3-гидрокси-масляная кислота, ацетоуксусная кислота и ацетон являются естественными конечными продуктами бета-окисления митохондриальных жирных кислот. Кетонурия, увеличение экскреции кетонов с мочой, обнаруживается физиологически в позднем младенчестве, детстве и подростковом возрасте, но не считается нормальным у новорожденных. Физиологический кетоз не сопровождается метаболическим ацидозом, гиперлактатемией или гипогликемией — маркерами метаболического стресса. Это обычное явление после голодания, рвоты, употребления кетогенной диеты или повышенного уровня катаболизма. Однако, поскольку кетоны являются органическими кислотами, тяжелая кетонурия, которая вызывает метаболический ацидоз, не должна рассматриваться как физиологическая и указывать на врожденные нарушения метаболизма. Время кетонурии в связи с кормлением или голоданием является важным фактором, который может указывать на потенциальный тип основного метаболического расстройства. Например, тяжелая кетонурия с гипогликемией натощак или постпрандиальная гипергликемия с гиперлактатемией являются общими признаками при некоторых нарушениях гликогеноза.
В отличие от других маркеров метаболического стресса, кетоз является клинически значимым как при повышении, так и при его отсутствии. В то время как серьезное снижение экскреции кетонов наряду с низким уровнем циркулирующей глюкозы (гипокетотическая гипогликемия) является обычным явлением при рвоте, анорексии или генерализованных катаболических состояниях, этот паттерн также является существенным индикатором потенциального нарушения окисления митохондриальных жирных кислот, с или без чрезмерного использования глюкозы. Однако важно отметить, что некоторые нарушения окисления жирных кислот в митохондриях могут сопровождаться периодическими эпизодами кетонурии от легкой до тяжелой степени, когда пораженный фермент достаточно дистален в пути деградации бета-окисления, что метаболизм длинноцепочечных жирных кислот все еще способен генерирование некоторых кетонов, как в случае дефицита 3-гидроксиацил-СоА-дегидрогеназы (HAD) или дефицита со средней цепью ацил-КоА-дегидрогеназы (MCAD).
Нейровизуализация
Помимо исследований, перечисленных выше, другие важные начальные тесты включают CBC, функциональные тесты печени, исследования коагуляции, уровни креатинкиназы, тесты почечной функции, BUN, исследование мочевой кислоты, липидных профилей и клеток CSF и глюкозу CSF.
Исследования нейровизуализации, включая МРТ и МР-спектроскопию, могут показать специфические изменения мозга, характерные для определенных наследственных дефектов, таких как симметричные базальные ганглии и вовлечение таламуса, наблюдаемые при митохондриальных расстройствах, субдуральных выпотах и сниженной оперкуляризации, наблюдаемой при глютариновой ацидемии типа I (GA1), отсутствующий пик креатина МР спектроскопия демонстрирует врожденные нарушения метаболизма креатина и изменения белого вещества при таких нарушениях, как Х-сцепленная адренолейкодистрофия и метахроматическая лейкодистрофия.
Лабораторные тесты
Лабораторные тесты, которые представляют широкую сеть и могут использоваться для диагностики множественных IEM, включают анализ мочи на органическую кислоту, и аминокислоты в плазме и моче, ацилкарнитин в плазме и анализ жирных кислот с очень длинной цепью в сыворотке. Поскольку вторичный дефицит карнитина, являющийся следствием потери карнитина в виде сложных эфиров ацилкарнитина в моче, является распространенным явлением при многих нарушениях обмена веществ, включая нарушения окисления жирных кислот и органические ацидурии, при оценке состояния пациента на предмет потенциального нарушения обмена веществ следует также включать количественное определение содержания карнитина в сыворотке.
Некоторые расстройства приводят к увеличению циркулирующих промежуточных метаболитов, которые, благодаря своему характеру, образуют основу для диагностики и диетического мониторинга (т.е. повышенный уровень фенилаланина в плазме и пониженный уровень тирозина при фенилкетонурии, повышенное содержание свободных жирных кислот при расстройствах окисления жирных кислот или повышенное число разветвленных цепей). аминокислоты при MSUD); другие расстройства приводят к повышенной экскреции ключевых метаболитов, которые могут использоваться для тех же целей (например, повышенный цистин мочи при цистинурии), в то время как некоторые расстройства вызывают несколько изменений. Увеличение содержания аномальных метаболитов в плазме также могут быть обнаружены и в моче, если концентрация крови достигает почечного порога для того или иного аналита. В целом, нарушения метаболизма аминокислот, которые влияют на стадии проксимального метаболизма (например, фенилкетонурия или MSUD), приводят к аномальным аминокислотным профилям плазмы.
Нарушения метаболизма аминокислот, которые влияют на дистальные ферментативные стадии (то есть изовалериановая ацидемия или глутаровая ацидурия типа I), могут или не могут иметь отклонения в анализе аминокислот в плазме, но, как правило, приводят к аномальным профилям мочевой органической кислоты. Нарушения, которые влияют на переносчики аминокислот в почках (например, цистинурия или непереносимость белка лизинурия), обычно лучше всего диагностируются по аминокислотному профилю мочи.
Состояния, влияющие на различные стадии общего метаболического пути, могут приводить к аномальным паттернам общих промежуточных метаболитов; в таких случаях может потребоваться дальнейшее тестирование, чтобы сузить диагноз, например, когда в плазменном профиле ацилкарнитина обнаружен повышенный уровень 3-гидроксиизовалерил- / 2-метил-3-гидроксибутирилкарнитина (C5OH). Этот аналит обнаружен при нескольких нарушениях, влияющих на метаболизм аминокислот с разветвленной цепью. В таких случаях может потребоваться анализ мочи на органическую кислоту или дополнительные биохимические или молекулярные анализы для подтверждения первоначальных результатов или для постановки окончательного диагноза.
В некоторых других условиях анализ активности фермента и / или молекулярный анализ могут быть необходимы для конкретного диагноза; следует отметить, что некоторые ферментные тесты требуют инвазивных процедур, таких как биопсия печени при заболевании хранения гликогена в печени или биопсия кожи при культивировании фибробластов для исследований окисления жирных кислот.
Категория сообщения в блог:
МИНОБРНАУКИ РОССИИ
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ОБРАЗОВАНИЯ
«НИЖЕГОРОДСКИЙ ГОСУДАРСТВЕННЫЙ ПЕДАГОГИЧЕСКИЙ УНИВЕРСИТЕТ ИМЕНИ КОЗЬМЫ МИНИНА»
Факультет психологии и педагогики
Кафедра специальной педагогики и психологии
Направление подготовки: 44.03.03 Специальное (дефектологическое) образование
Профиль подготовки: Олигофренопедагогика
Реферат
На тему: Наследственные заболевания обмена веществ как причина сложных дефектов.
Билялова Динара Рамильевна
Нижний Новгород – 2020 г
Оглавление
Вступление 3
Глава 1. Теоретический обзор проблемы наследственных заболеваний обмена веществ как причина сложных дефектов. 4
1.1. Наследственные заболевания 4
1.2. Сложные дефекты 6
1.3. Наследственные заболевания обмена веществ как причина сложных дефектов. 8
Глава 2. Практическая часть. 9
2.1. Основные формы НБО. 9
2.2. Классические неврологические синдромы, ассоциированные с известными НБО. 10
2.3. Преимущества генетического исследования заболеваний 11
2.4. Лечение 12
Заключение 13
Список используемой литературы: 15
Приложения 16
Вступление
Большинство наследственных заболеваний обмена веществ (также называемых врожденными дефектами метаболизма) обусловлены мутациями в генах, кодирующих ферменты; дефицит фермента или его неактивность приводят к накоплению субстрата или метаболитов, или к недостатку производных фермента. Такие заболевание являются актуальными в современном мире, потому что идентифицированы сотни таких расстройств, и, хотя большинство наследственных нарушений обмена веществ крайне редки по отдельности, в целом они представляют довольно распространенную группу расстройств.
Наследственные метаболические нарушения обычно группируются в зависимости от пораженного субстрата, например:
-
Нарушения обмена аминокислот
-
Нарушения углеводного обмена
-
Нарушения метаболизма жирных кислот
-
Нарушения метаболизма пуринов и пиримидинов
В большинстве стран обязательным является проведение неонатального скрининга всех новорожденных на наличие конкретных наследственных нарушений обмена веществ и других состояний, включая фенилкетонурию, тирозинемию, недостаточность биотинидазы, гомоцистинурию, болезнь кленового сиропа и галактоземию. Во многих государствах имеются расширенные скрининговые программы, которые охватывают множество врожденных нарушений мfетаболизма, включая нарушения окисления жирных кислот и другие органические ацидемии. Исчерпывающий обзор каждого из этих состояний см. в техническом отчёте «Листы и таблицы алгоритмов и аналитика состояния скрининга новорожденных» (newborn screening ACT sheets and algorithm table) Американской коллегии медицинской генетики и геномики (ACMG).
Нарушения обмена веществ, в основном вызывающие заболевания у взрослых (например, подагра, порфирия), органоспецифичные заболевания (например, болезнь Вильсона, врожденная гиперплазия надпочечников) или являются общими (например, кистозный фиброз, гемохроматоз), обсуждаются в других разделах Руководства. Для наследственных заболеваний метаболизма липопротеинов – Генетическая (Первичная) Дислипидемия.
Глава 1. Теоретический обзор проблемы наследственных заболеваний обмена веществ как причина сложных дефектов.
- Наследственные заболевания
Наследственные нарушения обмена веществ включают в себя большую группу наследственных заболеваний, затрагивающих расстройства метаболизма. Такие нарушения составляют значительную часть группы метаболических расстройств (метаболические заболевания).
Метаболические заболевания — гетерогенная группа болезней, при которых нормальные метаболические процессы в тканях нарушены, чаще всего из-за отсутствия или недостаточности определённого фермента, и, как следствие, патологического накопления веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений.
Типичные симптомы полиморфны и зачастую вовлекают несколько систем организма:
— Задержка роста, психо-моторного развития, потеря веса
— Недоразвитые гениталии, задержка или преждевременное половое созревание
— Задержка развития, судороги, деменция, энцефалопатия, инсульты
— Глухота, слепота, нарушение восприятия боли
— Кожная сыпь, гиперпигментация, недержание пигмента, избыточное оволосенение
— Аномалии развития зубов
— Иммунодефицит, тромбоцитопения, анемия, увеличенная селезенка, увеличенные лимфоузлы
— Повторяющаяся рвота, диарея, абдоминальные боли
— Повышение диуреза, нарушения функции почек, дегидратация, отеки
— Гепатомегалия, нарушение функции печени
— Малые аномалии развития (стигмы дизэмбриогенеза), врожденные пороки развития
— Тахипноэ, нарушения дыхания
— Нарушение поведения, депрессия, психозы
— Боль в суставах, мышечная слабость, крампи
В большинстве случаев врожденные нарушения метаболизма проявляются в первые дни жизни. Однако, они могут остаться нераспознанными в период новорожденности, и диагноз может быть поставлен только через несколько месяцев и даже лет, или же в редких случаях дебютировать в более позднем возрасте.
Врожденное нарушение обмена веществ должно рассматриваться, как возможное состояние, у любого ребенка с одним или более из указанных клинических проявлений:
1) необъяснимое отставание умственного, двигательного развития, судороги;
2) повышенный уровень определенных метаболитов в крови или моче (например, при исследованиях мочи на органические ацидурии или крови с помощью тандемной масс-спектрометрии)
3) необычный запах, в частности во время острого заболевания;
4) интермиттирующие эпизоды необъяснимой рвоты, ацидоза, нарушений психики, кома;
5) гепатомегалия;
6) почечные камни.
В данную панель входят такие группы заболеваний как:
— нарушения обменов углеводов (например, болезни накопления гликогена),
— аминоацидопатии (например, фенилкетонурия),
— органические ацидурии (например, алькаптонурия),
— нарушения окисления жирных кислот (например, глютаровая ацидемия 2 типа),
— митохондриальные болезни (например, синдром Кернс-Сейера),
— порфирии (например, острая перемежающаяся порфирия),
— нарушение пуринов или пиримидинов (например, синдром Синдром Лёша-Найхана),
— нарушение обмена стероидов (например, врожденная гиперплазия надпочечников),
— пероксисомные болезни (например, синдром Цельвейгера)
— лизосомные болезни (например, болезнь Гоше),
— и другие заболевания со схожими проявлениями.
- Сложные дефекты
Сложный дефект представляет собой не просто сочетание (сумму) двух и более дефектов развития; он является качественно своеобразным и имеет особую структуру, отличную от составляющих его аномалий.
Категорию детей со сложными дефектами составляют (42):
Дети с умственной отсталостью, отягощенной нарушениями слуха;
Дети с умственной отсталостью, осложненной нарушениями зрения;
Дети глухие слабовидящие;
Слепоглухонемые дети;
Дети с задержкой психического развития, которая сочетается с дефектами зрения или слуха;
Глухие дети с нарушениями соматического характера (врожденные пороки сердца, заболевания почек, печени, желудочно-кишечного тракта).
Кроме того, в дефектологической практике встречаются дети с множественными дефектами. К ним относятся:
1. Дети с умственной осталостью слепоглухие;
2. Дети с нарушениями опорно-двигательного аппарата в сочетании с дефектами органов слуха, зрения, речи или интеллектуальной недостаточностью.
Таким образом, к детям со сложными дефектами можно отнести детей, у которых отмечаются нарушения развития сенсорных и моторных функций в сочетании с недостатками интеллекта (задержка психического развития, умственная отсталость).
Причинами возникновения сложных дефектов могут быть, как указывает Б.П.Пузанов, наследственные и экзогенные факторы.
Наиболее тяжелой группой детей со сложными дефектами выступают слепоглухонемые дети. Её составляют дети, не только полностью лишенные зрения, слуха и речи, но и с парциальным (частичным) поражением сенсорной сферы: слепые с такой потерей слуха, которая препятствует усвоению речи на слух, и глухие с такой потерей зрения, которая препятствует зрительной ориентировке.
Специфической особенностью таких детей является практически полная невозможность получать информацию об окружающем по естественным каналам, что увеличивает значимость коррекционного образования для них, по сравнению с другими детьми, имеющими сложные дефекты. При этом у слепоглухонемого ребенка часто могут быть развиты все усложняющиеся формы общения — от элементарных жестов (воспринимаемых посредством осязания) до вербальной речи. Это позволяет таким детям относительно успешно овладевать программой средней общеобразовательной школы, а некоторым оканчивать высшие учебные заведения.
1.3. Наследственные заболевания обмена веществ как причина сложных дефектов.
Патологии, также известные как врожденные нарушения метаболизма, — это заболевания, причина которых кроется в генетическом изменении белка или фермента, в результате чего блокируется определенный процесс метаболизма. Такая блокировка влияет на нормальное функционирование некоторых клеток и органов и проявляется рядом симптомов, различных у каждого пациента. Среди таких симптомов могут встречаться разные виды неврологических синдромов.
Эта группа патологий очень обширна, однако ее можно систематизировать с помощью действующей классификации, которая в данный момент претерпевает значительные изменения ввиду того, что сегодня мы располагаем гораздо большими знаниями о базовых механизмах развития таких патологий. Ниже приведены основные группы патологий, составленные на основании типа поражения организма при каждой из них.
Врожденное нарушение метаболизма малых молекул:
-
Влияют на промежуточный метаболизм. Сюда входят аминоацидопатии (фенилкетонурия и пропионовая ацидурия). Также сюда входит нарушения обмена углеводов или нейромедиаторов и нейромодуляторов.
-
Врожденное нарушение энергетического обмена
-
Характеризуется недостаточной выработкой и использованием энергии. Сюда входят митохондриальные заболевания, недостаточная выработка пирувата или глюкозы (в мышцах или печени) и т. д.
Врожденное нарушение метаболизма сложных молекул:
Группа заболеваний, которые препятствуют синтезу больших молекул. Они проявляются в виде постоянных симптомов, не связанных с питанием. Сюда входят лизосомные (мукополисахаридоз, олигосахаридоз, сфинголипидоз и т. д.), пероксисомальные (синдром Цельвегера, адренолейкодистрофия, сцепленная с хромосомой Х) заболевания и врожденные нарушения гликозилирования, а также другие врожденные нарушения метаболизма.
Глава 2. Практическая часть. 2.1. Основные формы НБО.
Мутации в отдельных генах приводят к нарушению синтеза или разрушения белков, углеводов, жиров или сложных веществ. Большинство из них связаны с дефектами ферментов или транспортных белков, в результате чего происходит блок определенного метаболического пути, определенная биохимическая реакция перестает работать и в клетках накапливаются субстраты этих реакций и их производные.
Основная симптоматика проявляется из-за накопления токсических веществ перед блоком, патологических альтернативных путей метаболизма, уменьшения продукции энергетических субстратов или как следствие дефицита конечных продуктов биохимической реакции после блока метаболического пути.
Почти каждое заболевание из этой группы имеет несколько форм, которые различаются по возрасту начала заболевания, клинической выраженности, и, нередко, по типу наследования.
-
Нарушения обменов углеводов (например, болезни накопления гликогена)
-
Аминоацидопатии (например, фенилкетонурия)
-
Органические ацидопатии (например, алькаптонурия)
-
Нарушения окисления жирных кислот (например, глютаровая ацидемия 2 типа)
-
Лизосомные болезни (например, болезнь Гоше)
-
Порфирии (например, острая перемежающаяся порфирия)
-
Нарушение пуринов или пиримидинов (например, синдром Синдром Лёша-Найхана)
-
Нарушение обмена стероидов (например, врожденная гиперплазия надпочечников)
-
Пероксисомные болезни (например, синдром Цельвейгера)
-
Митохондриальные болезни (например, синдром Кернс-Сейера)
2.2. Классические неврологические синдромы, ассоциированные с известными НБО.
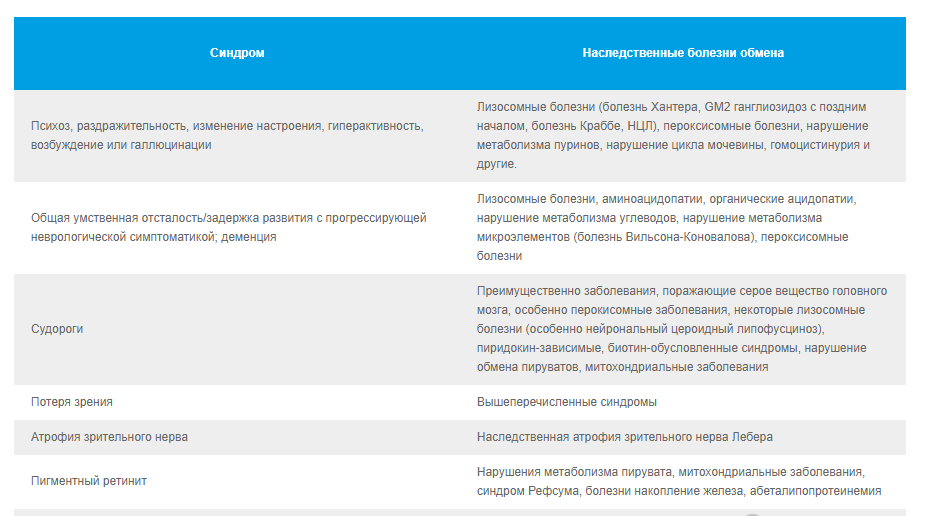
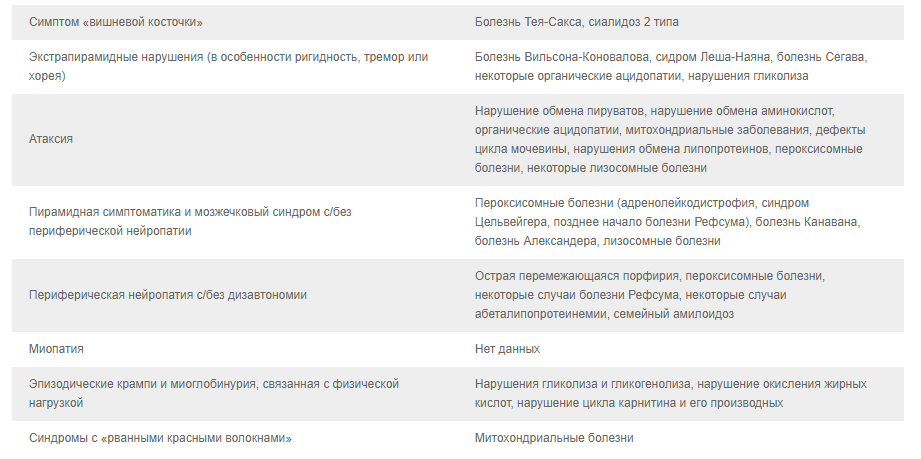
2.3. Преимущества генетического исследования заболеваний
![]()
Генетические исследования позволяют выявлять мутации в известных генах, ассоциированных с НБО. Со временем уже существующие панели генов дополняются и расширяются с учетом мировых баз данных.
![]()
Точный молекулярно-генетический анализ позволяет не проводить дальнейшие диагностические исследования и более точно прогнозировать течение заболевания.
![]()
Анализ проходит быстро и безболезненно. Всё, что требуется для анализа – это образец крови. Специалисты сравнивают результаты со специальной панелью, которая позволяет выявить возможную причину заболевания.
![]()
Пациент получает точные результаты, на основании которых специалисты могут назначить максимально эффективное лечение.
![]()
При проведении процедуры строго соблюдается конфиденциальность.
2.4. Лечение
Большинство наследственных болезней обмена в настоящее время являются неизлечимыми, и терапевтические возможности сводятся к симптоматической и патогенетической терапии: диетотерапия (ограничение поступления в организм определенных веществ), специальные лечебные продукты питания, препараты, направленные на снижение образования токсичных метаболитов, восполнение некоторых метаболитов, которых не хватает в организме, фермент-заместительная терапия.
При некоторых формах НБО (мукополисахаридоз 1 типа, ряд анемий и наследственных нарушениях иммунной системы) эффективно проведение трансплантации гемопоэтических стволовых клеток.
Поэтому важно установить диагноз как можно раньше, чтобы лечение было более эффективно.
Заключение
Исходя из всего выше сказанного, можно сделать вывод о том, что наследственные нарушения обмена веществ включают в себя большую группу наследственных заболеваний, затрагивающих расстройства метаболизма. Такие нарушения составляют значительную часть группы метаболических расстройств (метаболические заболевания). Наследственными называются такие заболевания человека, которые вызваны перестройками и нарушениями в генетическом материале организма – хромосомах и генах. Среди наследственных заболеваний человека одно из самых значительных мест занимают наследственные болезни обмена. В настоящее время эта группа включает около 700 различных заболеваний
Развитие большинства из них является следствием дефекта единичных генов, кодирующих индивидуальные ферменты, которые обеспечивают превращение одних веществ (субстраты) в другие (продукты). В большинстве случаев таких расстройств патогенным является накопление веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений. Для наследственных болезней обмена веществ иногда используется синонимичный термин «врождённые ошибки метаболизма» ферментопатии.
Список используемой литературы:
-
Talking glossary of genetic terms: genome (англ.). National Human Genome Research Institute. Дата обращения 1 ноября 2012.Архивировано 4 ноября 2012 года.
-
↑ International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. (англ.) // Nature : journal. — 2004. — Vol. 431, no. 7011. — P. 931—945. [1]
-
↑ Перейти обратно:1 2 International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. (англ.) // Nature : journal. — 2001. — Vol. 409, no. 6822. — P. 860—921. [2]
-
↑ «Мусорная» ДНК помогает включать гены.
-
↑ «Мусорная» ДНК играет важнейшую роль в поддержании целостности генома.
-
↑ Tjio J. H., Levan A. The chromosome number of man (неопр.) // Hereditas (англ.)русск.. — 1956. — Т. 42. — С. 1—6. Первая работа с точно установленным числом хромосом у человека.
-
↑ Human Chromosome Number, здесь рассказана история подсчёта хромосом у человека
-
↑ Nei M., Xu P., Glazko G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 2001. — Vol. 98, no. 5. — P. 2497—2502.
Приложения
ПРИЛОЖЕНИЕ 1

ПРИЛОЖЕНИЕ 2

ПРИЛОЖЕНИЕ 3
Биохимические
особенности организма определяют
приблизительно 50 000 пар генов,
передаваемых из поколения в поколение
через хромосомы. Индивидуальные вариации
возникают в результате случайного
отбора и рекомбинации во время
редукционного деления (мейоза), а также
в результате периодических мутаций.
Последствия таких вариаций бывают
различными: от изменении, несовместимых
с жизнью, до возникновения биохимических
особенностей, которые, если и удается
обнаружить, то лишь с помощью
специальных методов исследования. К
этой последней категории относятся
генетические вариации таких белков
плазмы крови, как трансферрины и
гаптоглобины, изменения которых важны
в связи с популяционными и генетическими
исследованиями, но не всегда приводят
к функциональным расстройствам. Наряду
с двумя указанными выше крайними
вариантами существует множество
промежуточных вариаций, приводящих к
развитию функциональных аномалий, для
обозначения которых и применяют
термин врожденные нарушения обмена
веществ.
ОБЩИЕ
ПРИНЦИПЫ
Поскольку
последовательность оснований в нитях
ДНК, представляющих собой гены,
кодирует через РНК структуру белков,
не удивительно, что большинство (если
не все) биохимических аномалий можно
объяснить нарушениями биосинтеза
одного пептида. Такие аномалии могут
быть обусловлены наличием либо
измененного структурного гена,
кодирующего образование вариантного
белка, либо измененного регуляторного
гена, что приводит к нарушению
функционирования одного или нескольких
структурных генов и, следовательно, к
вариациям содержания одного или
нескольких белков с неизмененной
структурой. В большинстве примеров,
которые мы рассматриваем в этой главе,
измененные белки являются ферментами.
Возможные
последствия для обмена веществ
Последствия
недостаточности одного фермента в цепи
реакций обмена веществ могут проявляться
поразному. Предположим, что превращение
соединения А в соединение Б катализирует
фермент Е и что соединение В встречается
на альтернативном пути превращений
(рис. 41).
Последствиями
недостаточности Е могут быть следующие
явления: 1) недостаточность продукта
ферментативной реакции (Б). В качестве
примеров укажем на недостаточность
кортизола при врожденной гиперплазии
надпочечников и на гипогликемию при
некоторых формах гликогенозов;
Рис.
41. Схема альтернативных путей
биохимических превращений.

2)
накопление вещества, превращение
которого катализирует фермент (А)
(например, фепилаланин при фенилкетонурии).
При многих лизосомных болезнях
накопления, вещества, в норме подвергающиеся
гидролизу в лизосомах, накапливаются
в них в связи с недостаточностью одного
из ферментов; 3) отклонение на альтернативный
путь с образованием некоторых
биологически активных соединений (В).
К этой группе явлений относится
вирилизация, обусловленная андрогенами,
при врожденной гиперплазии надпочечников.
Если
метаболическое превращение в целом
регулируется по принципу обратной
связи конечным продуктом, то эффекты
двух последних типов аномалий будут
особенно значительными. Так, например,
при врожденной гиперплазии надпочечников
недостаточность кортизола стимулирует
синтез стероидов и, следовательно,
накопление андрогенов, в результате
чего развивающаяся вирилизация еще
более усиливается.
Факторы
внешней среды могут модифицировать
(или даже полностью определять)
клинические проявления некоторых
врожденных нарушений обмена веществ.
Так, например, поскольку у женщин во
время менструации и беременности
происходит потеря организмом железа,
при идиопатическом гемохроматозе у
женщин накапливается меньше железа,
чем у мужчин при этом же заболевании.
У больных с вариантными формами
холинэстеразы длительный паралич
развивается только после введения
миорелаксанта дитилина (с. 386); у некоторых
больных с недотаточностью
глюкозо6фосфатдегидрогеназы гемолиз
начинается только после приема внутрь
таких лекарственных средств, как
примахин. Такие больные представляются
«здоровыми» в отсутствие контактов с
современными лекарственными средствами.
Когда
возникает предположение о наличии
врожденного нарушения метаболизма
Возможность
врожденного нарушения обмена веществ
следует предполагать, если в раннем
или позднем детском возрасте обнаруживают
клинические или биохимические аномалии
(особенно у двух пли большего числа
детей в одной семье). Следующие явления
заслуживают особого внимания в этой
связи (если для их возникновения нет
очевидных причин): 1) нарушение развития;
2)
рвоты; 3) гипогликемия; 4)’ особый запах
или окраска пеленок;
5)
гепатоспленомегалия; 6) желтуха; 7)
задержка умственного развития, припадки,
спастическое состояние мышц; 
метаболический ацидоз; 9) почечные
камни; 10) рахит, не поддающийся лечению.
Клиническое
значение врожденных нарушений метаболизма
Выявление
многих врожденных нарушений обмена
веществ представляет только академический
интерес, так как клинически они не
проявляются. В тех случаях, когда не
разработаны эффективные методы
лечения, может оказаться важной
диагностика как основа проведения
генетического консультирования.
Распознавание ряда заболевании в
рапном детским возрасте жизненно важно,
поскольку лечение может предотвратить
развитие необратимых клинических
явлений и гибель больного. Среди наиболее
важных заболеваний такого типа назовем
фенилкетонурию, галактоземию, болезнь
«моча с запахом кленового сиропа».
При
ряде заболеваний показаны обследования
родственников больного как для
предупреждения дальнейшего развития
заболевания, так и в связи с
необходимостью избегать воздействия
усугубляющих патологическое состояние
факторов. Примерами патологических
состояний такого типа являются аномалии
холинэстеразы, недостаточность
глюкозо6фосфатдегидрогеназы, острые
порфирии, гемохроматоз, цистинурия,
болезнь Уилсона.
Примерами
патологических состояний, поддающихся
симптоматическому лечению, являются
врожденный нефрогенньш несахарный
диабет, врожденная недостаточность
дисахаридазы, болезнь Хартнапа.
Некоторые
врожденные нарушения обмена веществ
полностью (или почти полностью) безвредны.
Они важны в том смысле, что могут
приводить к диагностическим ошибкам
или напрасно беспокоить пациента.
Примерами таких состояний являются
ренальная гликозурия, алкаптонурия,
болезнь Жильбера.
Наконец,
некоторые врожденные нарушения обмена
веществ могут клинически проявиться
только после достижения половой
зрелости. В этих случаях желательна
генетическая консультация кровных
родственников больного. Примером
заболевания такого типа может быть
болезнь Уилсона.
Лабораторная
диагностика врожденных нарушений
метаболизма
О
недостаточности фермента обычно судят
косвенно по повышению концентрации
исходного вещества, которое в норме
подвергается биохимическим превращениям,
катализируемым данным ферментом
(например, фенилаланин при фенилкетонурии).
Прямые определения активности таких
ферментов проводят только в
специализированных центрах, но по
возможности диагноз во всех случаях
следует подтверждать этим методом.
Пренатальная диагностика некоторых
врожденных нарушений метаболизма
возможна путем исследований клеток
амниотической жидкости, полученных
на ранних стадиях беременности и
клуьтивируемых
in vitro.
Скрининг
для выявления врожденных нарушений
метаболизма
Учитывая
важность раннего проведения лечения,
многие страны утвердили программы
скрининга всех новорожденных для
выявления врожденных нарушений
метаболизма, особенно фенилкетонурии.
Для тестирования можно использовать
кровь (полученную после прокола кожи
на пятке) или мочу. Кровь (или в более
редких случаях мочу) часто собирают на
фильтровальную бумагу; в таком виде ее
легко доставлять в лабораторию.
Важно
учитывать время отбора проб для анализов,
чтобы избежать получения
ложноотрицательных результатов.
Соединения, лежащие на путях метаболизма
выше точки блокировки ферментативных
превращений (например, фенилаланин при
фенилкетонурии или галактоза при
галактоземии), накапливаются только
после того, как ребенок начинает получать
с пищей соответствующие предшественники
(такие как белковые или молочные
продукты). При скрининговом обследовании
детей, которых считают здоровыми,
кровь обычно берут на 6—9й день жизни.
Аномальные метаболиты могут быть не
определены в моче до 4—6 нед после
рождения, если почечный порог для них
относительно высок.
Полученный
при скрининге положительный результат
следует подтвердить путем количественного
анализа или повторного тестирования.
Многие обнаруживаемые аномалии являются
транзиторными.
В
конце этой главы мы приводим ссылку на
статью, в которой обсуждаются типичные
ошибки, допускаемые при интерпретации
результатов скринингового обследования
новорожденных с целью выявления
врожденных нарушений метаболизма, и
указывается на ответственность
исследователей, проводящих такую
работу.
Лечение
при врожденных нарушениях метаболизма.
Некоторые врожденные нарушения
метаболизма поддаются лечению путем
доставки в организм недостающего
метаболита или путем ограничения
поступления в пищеварительный тракт
предшественников нарушенных процессов
обмена веществ. Иногда могут быть
удалены накапливающиеся продукты
(например, железо при гемохроматозе).
Характер
наследования
Следующий
раздел представляет собой только
краткий обзор; подробное изложение
вопроса имеется в книгах по генетике.
Любой
наследуемый признак определяет пара
генов на гомологичных хромосомах (по
одной от каждого из родителей). Аллелями
называют различные гены, определяющие
один и тот же признак. Индивидуум,
обладающий двумя идентичными аллелями
является гомозиготным в отношении
данного гена или наследуемого
признака; если он имеет два различных
аллеля, то он гетерозиготный. Носителями
генов могут быть половые хромосомы (х
и у) или аутосомы (сходные у представителей
обоих полов); при этом характер
наследования различен.
Аутосомное
наследование
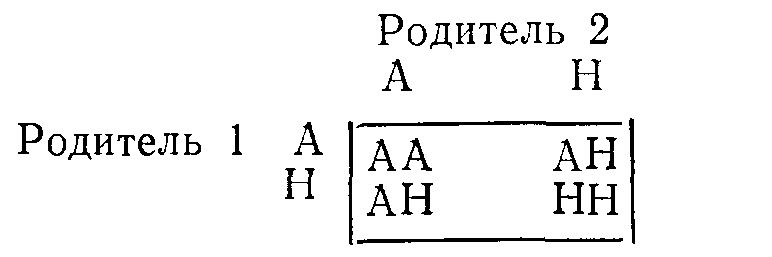
1.
Предположим, что один из родителей
(родитель 1 в приведенном ниже примере)
является носителем аномального гена
(А). Если Н обозначает нормальный ген,
то возможности комбинаций генов у
потомства показаны в квадрате.

Видно,
что на основе данных статистики половину
потомков следует считать носителями
одного аномального гена (АН); они будут
гетерозиготными в отношении этого
гена, как и родитель 1. Ни один из потомков
не будет гомозиготным в отношении
аномального гена (АА).
2.
Если оба родителя гетерозиготны, то
четверть их потомков (при общем большом
числе) будет гомозиготна (АА) и половина
гетерозиготна.
3.
Если один родитель гомозиготен, а другой
нормален, то все потомки будут
гетерозиготными.
Поскольку
гены, определяющие клинически
диагностируемые аномалии, как правило,
встречаются редко, приведенный выше
пример 1 статистически наиболее вероятен.
При браках между родственниками пример
2 становится более вероятным, так как
среди кровных родственников более
вероятна возможность наличия
носителей одинаковых аномальных генов,
чем среди людей, не являющихся
родственниками.
Последствия
носительства аномального гена зависят
от его относительной мощности по
сравнению с нормальным.
Доминантный
ген вызывает аномалии как у гетерозиготных,
так и гомозиготных носителей, хотя
степень поражения может быть более
высокой у гомозигот. Так, в примере 1
будут поражены родитель 1 и половина
потомков, а в примере 2 — оба родителя
и 75% потомков. Характерно проявление
аномалий в последующих поколениях.
Рецессивный
ген вызывает аномалии только у
гомозиготных индивидуумов. Так, в
примере 1 ни родители, ни потомки не
будут поражены, а в примере 2 родители
будут казаться здоровыми, но 25% потомков
будут поражены. Характерно проявление
одного или нескольких случаев заболевания
лишь в одном поколении (при клинически
здоровых родителях).
Понятия
доминантный и рециссивный относительны.
Доминантный ген может не проявлять
себя (неполная пенетрантность) и таким
образом как бы «пропускать» поколение.
Различной может быть степень
экспрессии гена и, следовательно,
степень выраженности аномалии,
которую он определяет. Наконец,
рецессивный ген, вызывающий заболевание
только у гомозигот, может быть тем не
Meiiee обнаружеи
с помощью биохимических тестов п у
гетерозиготных носителей.
Наследование,
сцепленное с полом
Для
некоторых аномальных генов носителями
являются только половые хромосомы,
почти всегда Ххромосомы.
Х-сцепленное
рецессивное наследование.
Женщины являются носителями двух
Ххромосом, а мужчины — одной Х и одной
Y. При Хсцепленном
рецессивном наследовании аномальная
Ххромосома (Ха) находится в латентном
состоянии, если она сочетается с
нормальной Ххромосомой, но становится
активной при сочетании с Yхромосомой.
Если мать является носительницей Ха,
она будет клинически здоровой, но
половина (статистически) ее сыновей
будут поражены
(YXa). Половина
дочерей будут носительницами (ХХа),
но клинически все дочери будут здоровыми.

Если
отец поражен заболеванием и мать
является носительницей двух нормальных
генов, ни один из сыновей не будет
поражен, но все дочери будут
гетерозиготными носительницами.

Для
Х-сцепленного рецессивного наследования
характерно проявление врожденных
заболеваний только у потомков мужского
пола, тогда как гетерозиготными
носительницами являются женщины.
Клинические симптомы заболевания у
женщин встречаются редко, только
когда они гомозиготны по аномальному
гену.
Такая
ситуация может возникать в тех случаях,
если поражен отец, а мать является
гетерозиготной носительницей.
Классическим
примером Хсцепленного рецессивного
наследования является гемофилия.
Х-сцепленное
доминантное наследование.
При этом типе наследования поражены
как ХХа, так и YXa (женщины и мужчины),
например, при врожденной гипофосфатемии.
Множественные
аллели. В
некоторых случаях один и тот же признак
могут определять несколько аллелей.
Разные сочетания пар могут при этом
приводить либо к развитию различных
заболеваний (например, некоторых
гемоглобинопатий), либо к возникновению
вариации, которые удается обнаружить
только при биохимическом тестировании
(например, вариации белков плазмы
крови).
ЗАБОЛЕВАНИЯ,
ОБУСЛОВЛЕННЫЕ ВРОЖДЕННЫМИ НАРУШЕНИЯМИ
МЕТАБОЛИЗМА
Мы
рассмотрим лишь очень немногие из числа
изученных врожденных нарушений
метаболизма. Избранные нами примеры
иллюстрируют некоторые принципы,
упомянутые выше. Сделанный выбор,
безусловно, определяется нашими
представлениями об относительной
важности отдельных вопросов. С этими
представлениями, возможно согласятся
не все исследователи. На с. 391 были
перечислены некоторые аномалии,
представляющиеся наиболее важными для
клиники. В более полном (но, конечно, не
исчерпывающем) перечне, представленном
на с. 406, врожденные патологические
состояния систематизированы; по
возможности указав характер их
наследования. Многие из этих заболеваний
мы уже кратко упоминали в соответствующих
главах этой книги. Остается рассмотреть
лишь немногие патологические состояния,
которые, по нашему мнению, представляются
относительно более важными по сравнению
с другими, не упоминаемыми в тексте.
Частота
заболеваний.
Все врожденные нарушения метаболизма
встречаются очень редко. В ряде стран
в ходе осуществления программ скрининга
была приблизительно оценена частота
заболеваний, связанных с врожденными
нарушениями метаболизма, Из числа
заболеваний, рассматриваемых ниже,
фонплкетонурия, болезнь Хартнапа,
цистинурия, врожденная иминоглицинурия
в гистидинемия распространены особенно
широко (1 на 10000— 20 000); болезнь «моча с
запахом кленового сиропа» встречается
значительно реже (приблизительно 1 на
350 000).
Аминоацидурия
Поскольку
нарушения метаболизма или экскреции
аминокислот встречаются при многих
врожденных аномалиях обмена веществ,
аминоацидурия представляет собой один
из первых симптомов, который следует
искать в таких случаях.
В
норме аминокислоты фильтруются в
почечных клубочках и поступают в
проксимальные отдельг канальцев с
концентрацией, равной таковой в плазме;
почти все они здесь активно реабсорбируются.
Таким образом, возможна аминоацидурия
двух типов:
1)
аминоацидурия перегрузки, когда в связи
с повышением содержания аминокислот
в плазме крови они поступают в
проксимальный отдел канальцев в
концентрациях, превосходящих
реабсорбционную мощность клеток
почечных канальцев; 2) почечная
аминоацидурия, при которой содержание
аминокислот в плазме крови снижено в
результате потерь их из организма с
мочой вследствие недостаточности
реабсорбции в канальцах.
На
основании «данных о характере
экскретируемых с мочой аминокислот
можно различать два типа аминоацидурий:
1) специфическую аминоацидурию, когда
чрезмерно экскретируется либо одна
аминокислота, либо группа родственных
аминокислот. Такая аминоацидурия,
обусловленная как перегрузкой, так и
почечной недостаточностью, почти всегда
генетически предопределена;
2)
неспецифическую амипоацидурию, когда
происходит чрезмерная экскреция
целого ряда аминокислот неродственных
между собой.
Такая
аминоацидурия почти всегда является
не врожденной, а приобретенной. Она
может быть обусловлена перегрузкой
как, например, при тяжелых заболеваниях
печени, когда нарушение дезаминирования
аминокислот приводит к повышению их
содержания в плазме крови. Чаще
встречается почечная аминоацидурия,
связанная с неспецифическим поражением
проксимальных отделов канальцев. В
таких случаях, обозначаемых термином
синдром Фанкони, другие вещества,
реабсорбированные в проксимальных
отделах канальцев, также выводятся из
организма с мочой в чрезмерных
количествах (фосфоглюкоаминоацидурия).
При врожденных нарушениях метаболизма
развитие синдрома Фанкони значительно
чаще обусловлено вторичным повреждением
почечных канальцев веществами, не
подвергающимися нормальным биохимическим
превращениям (как, например, медь при
болезни Вильсона), чем первичным
генетическим дефектом.
Врожденные
аномалии механизмов транспорта веществ
Группы
химически родственных аминокислот
часто реабсорбируются в почечных
канальцах при участии одних и тех же
или взаимосвязанных механизмов. В
некоторых случаях аналогичные
группоспецифические механизмы участвуют
в процессах всасывания в кишечнике,
и при явлениях недостаточности бывают
поражены как почечные канальцы, так
и слизистая оболочка кишечника.
Идентифицированы врожденные нарушения
следующих группоспецифических
механизмов транспорта аминокислот:
1)
двухосновных (т. е. имеющих 2 аминогруппы
в молекуле) аминокислот цистина,
орнитина, аргинина и лизина (ЦОАЛ —
полезный мнемонический прием)
(цистинурия); 2) многих нейтральных
аминокислот (имеющих одну амино и одну
карбоксильную группу в молекуле)
(болезнь Харнапа); 3) иминокислот, пролина
и оксипролина, возможно, в сочетании с
глицином (врожденная иминоглицинурия).
Цистинурия
Цистинурия
обусловлена врожденной аномалией
реабсорбции в почечных канальцах
двухосновных аминокислот цистина,
орнитина, аргинина и лизина, что
приводит к чрезмерной экскреции с мочой
этих 4 аминокислот. Аналогичное нарушение
процессов транспорта аминокислот
возможно и в слизистой оболочке
кишечника, но, хотя всасывание цистина
и понижается, но синтезируется в
организме и его дефицит не развивается.
Недостаточность реабсорбции в почечных
канальцах приводит к высокой экскреции
цистина с мочой. Поскольку его
расгиордмосгь итносытельно невелика,
он может выпадать в осадок в мочевых
путях, что способствует образованию
камней со всеми последующими осложнениями.
Растворимость цистина такова, что
только у гомозигот его концентрации в
моче достигают уровня, при котором
возможны выпадение цистина в осадок,
кристаллурия и образование камней,
хотя повышенную экскрецию цистина с
мочой можно определить и у гетерозиготных
носителей.
Диагностика
цистипурии основана на обнаружении
чрезмерной экскреции с мочой цистипа
и других характерных аминокислот.
Данные о последних необходимы для того,
чтобы дифференцировать гомозигот,
в организме которых происходит
образование камней, от цистинлизинурий
у гетерозигот, а также от цистинурии,
составляющей часть генерализованной
аминоацидурий.
Лечение
при цистинурии направлено на предотвращение
образования камней путем введения
в организм днем и ночью больших объемов
жидкости, что приводит к снижению
концентрации цистина в моче.
Растворимость цистина повышается также
при подщелачивании мочи. Если эти меры
оказываются неадекватными, можно
попытаться вводить Dпеницилламин, в
присутствии которого образуется
более растворимое соединение.
Различные
генетические формы цистинурии наследуются
по аутосомнорецессивному типу. Многие
случаи бессимптомньге. Описанные выше
относительно безвредные явления
необходимо отличать от
цистиноза. Это
весьма редкое врожденное нарушение
обмена цистина характеризуется его
накоплением в клетках многих тканей.
В почках такое нарушение обмена приводит
к поражению канальцев и затем к
развитию синдрома Фанкони. Эта
аминоацидурия является неспецифической
и имеет почечное происхождение.
Больные умирают в раннем детском
возрасте.
Болезнь
Хартнапа
Болезнь
Хартнапа, названная по фамилии больного,
у которого она бьша впервые идентифицирована,
представляет собой редкое, во интересное
патологическое состояние, характеризуемое
нарушением транспорта нейтральных
аминокислот в почках и пищеварительном
тракте.
Большинство
(если не все) клинических симптомов
этой болезни может быть связано с
уменьшением всасывания в кишечнике
триптофана и с увеличением его экскреции
с мочой. В норме триптофан частично
превращается в никотинамид. Этот процесс
имеет особенно важное значение, если
поступление в организм никотинамида
с продуктами питания ограничено. На
этом примере можно видеть, как
изменения условий внешней среды
воздействуют на проявления врожденных
нарушений обмена веществ. Клинические
проявления болезни Хартнапа имеют
перемежающий характер и напоминают
симптомы пеллагры, а именно: 1) красная
чешуйчатая сыпь на открытых
поверхностях кожи; 2) обратимая мозжечковая
атаксия: 3) спутанность сознания,
выраженная в различной степени.
То,
что эти клинические симптомы обусловлены
недостаточностью никотинамида,
подтверждается данными о терапевтической
эффективности введения данного витамина
и наличием периода алиментарной
недостаточности, часто предшествующего
развитию проявлений заболевания.
Несмотря
на общую недостаточность процесса
всасывания аминокислот, признаки
обусловленных алиментарными факторами
нарушений обмена белков отсутствуют,
что, по-видимому, объясняется
всасыванием интактных пептидов
(очевидно, посредством других путей).
Сопутствующим
результатом лабораторных исследовании
является экскреция с мочой чрезмерных
количеств соединений индола. Эти
соединения образуются в кишечнике при
действии бактерий на неабсорбированный
триптофан. Болезнь Хартнапа наследуется
по рецессивному типу.
Диагностика
основана на обнаружении в моче
характерного сочетания экскретируемых
аминокислот. Существующие методы
исследования не позволяют легко
установить гетерозиготных носителей
данного заболевания.
Наследственная
иминоглицинурия.
Аномалия механизма трансапорта
аминокислот приводит к повышению
экскреции с мочой пролина, оксипролина
и глицина, по их содержание в плазме
крови остается в пределах нормы. Это
явление представляется безвредным,
но его необходимо отличать от других,
более серьезных. случаев иминоглицинурии.
Данное состояние наследуется по
аутосомнорецессивному типу.
Нарушения
обмена аминокислот
Описано
множество врожденных нарушений обмена
аминокислот. Большинство из них
характеризуется повышенным содержанием
соответствующих аминокислот в крови
и аминоацидурией, обусловленной
перегрузкой. Мы описываем в этом
разделе.’ лишь немногие, более подробно
изученные, нарушения.
Нарушения
обмена ароматических аминокислот
На
рис. 42
представлена схема основных химических
превращений этих аминокислот, а
также известных в настоящее время
нарушений активности ферментов,
катализирующих эти превращения.
Показано, что тирозин, который в норме
образуется в организме из фенилаланина,
является предшественником целого ряда
важных соединений.
Фенилкетонурия.
Данное патологическое состояние
вызывается аномалией системы
фенилалапингидроксилазы, обычно самого
фермента, но в некоторых случаях может
быть нарушен биосинтез его кофактора
тетрагидробиотерина. Таким образом,
весьма сходные между собой аномалии
биохимических процессов и клинические
симптомы могут быть обусловлены
несколькими различными врожденными
нарушениями метаболизма. Поскольку
фенилалашшгидроксилаза катализирует
иревращепие енилаланина в тирозин, при
рассматриваемом патологическом
состоянии в крови накапливается
феналаланин, который (в сочетании с
такими продуктами его побочного
превращения, как фенилпироиноградная
кислота) экскретируется с мочой. Этот
факт (экскреция фенилкетонов с мочой)
получил отражение в названии заболевание
широко практикуются скрининговые
исследования новорожденных с целью
раннего выявления случаев фенилкетонурии.
Проблемы, связанные с этими обследованиями,
мы обсудим ниже.
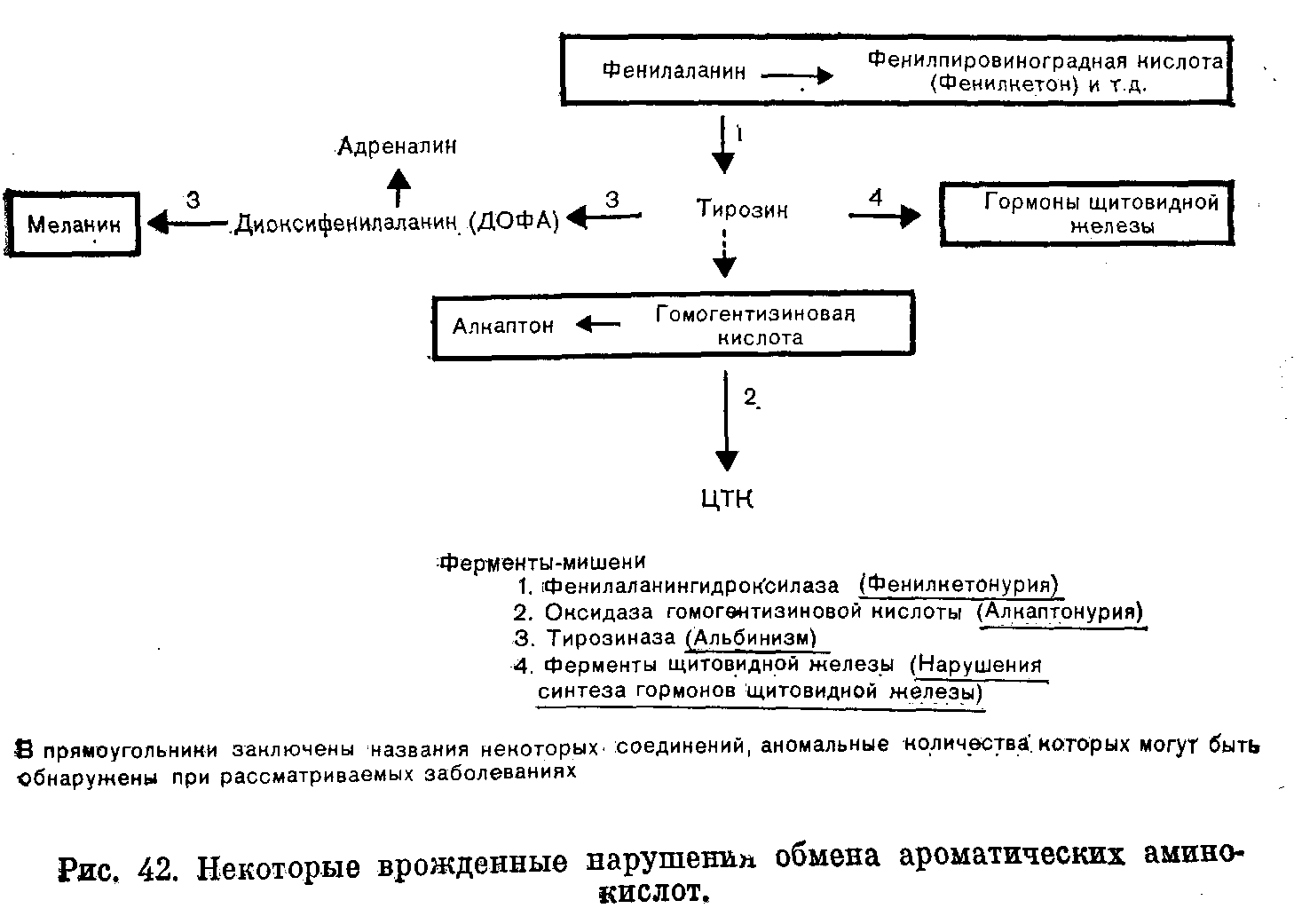
Отметим
следующие клинические симптомы этой
болезни:
1)
раздражительность, затруднения
при кормлении
новорожденных, рвоты и припадки в
течение первых нескольких недель
жизни; 2) задержка психического
развития в период между 4 и 6 мес жизни
на фоне необычной психомоторной
раздражительности 3) генерализованная
экзема (во многих случаях); 4) тенденция
к понижению меланинообразования. У
многих больных бледная кожа, светлые
волосы, голубые глаза.
Диагностика.
Концентрация фенилаланина может быть
измерена в крови, взятой из прокола
кожи на пятке. Эта методика пригодна
для массовых скрининговых обследований.
Лучше всего проверить тестирование в
специализированных центрах. Особенно
важное значение имеет время проведения
теста. Рекомендуется проводить
тестирование между 6 и 10 днями жизни
(т. е. наканупс выписки ребенка из
стационара).
Фенилпировиноградная
кислота мочи реагирует с хлорным
железом или препаратом фенистикс.
Однако такое тестирование может
дать положительный результат только
примерно через 6 нед; после рождения,
когда уже могут развиться необратимые
поражения головного мозга.
После
внедрения скрининговых тестов в
клиническую практику стала очевидной
возможность повышенного содержания
фенилаланина в крови не только при
фенилкетонурии, по и при других
состояниях, в частности у недоношенных
детей, что, по-видимому, обусловлено
замедлением созревания ферментных
систем. Такие случаи можно идентифицировать
путем повторных тестирований крови и
определения в крови содержания тирозина,
концентрация которого не повышена при
фенилкетонурии, но повышена при многих
других состояниях.
Описана
нетипичная стойкая (персистирующая)
гиперфенилаланинемия, не сопровождающаяся
задержкой умственного развития.
У
детей, которые
in utero подвергались
воздействию высоких концентраций
фенилаланина, если у их матерей была
недиагностированная или нелеченая
фенилкетонурия, могут быть признаки
умственной отсталости и других врожденных
аномалий, хотя сами по себе они не
страдают фенилкетонурией (материнская
фенилкетонурия).
Лечение.
Цель лечения — понижение уровней
фенилаланина в крови с помощью диеты,
бедной фенилаланином. Такое лечение
является трудным, дорогим и утомительным
для больных и их родителей; оно требует
тщательного биохимического мониторинга
концентрации фенилаланина в крови.
Недостаточность фенилаланина сама
по себе оказывает вредное воздействие.
В состав диеты необходимо включать
тирозин, являющийся предшественником.
многих важных продуктов обмена (см.
рис. 42).
Фенилкетонурия
наследуется по аутосомнорециссивному
типу. Гетерозиготные носители клинически
здоровы, но могут быть идентифицированы
путем биохимического тестирования.
Алкаптонурия.Врожденная недостаточность окспдазы
гомогентизиновой кислоты приводит к
алкаптонурии. Гомогентизиновая кислота
накапливается в крови, тканях и моче.
Окисление и полимеризация этого вещества
приводит к образованию пигмент»
алкаптона, подобно тому как полимеризация
ДОФА (см. рис. 42) приводит к образованию
меланина. Отложение алкаптона в хрящах,
которые затем темнеют, называют
охронозом. В последующие годы он может
вызвать артрит, а также заметное при
клиническом обследовании потемнение
ушей. Превращению гомогентизиновой
кислоты в алкаптон способствует щелочная
среда; при алкаптонурии наиболее
явным симптомом является экскреция
либо черной мочи, либо мочи, которая
темнеет по мере защелачивания при
хранении. Но во многих случаях этот
симптом может отсутствовать. Часто
первые признаки аномалии замечает
мать, обеспокоенная темным цветом
детских пеленок, которые становятся
еще темнее при стирке с применением
щелочного мыла или стиральных порошков.
Указанная аномалия совместима с
нормальной продолжительностью жизни
и не требует лечения, но в среднем
возрасте и позже обычно развивается
артрит. Гомогентизиновая кислота —
восстанавливающее вещество, реагирующее
с таблетками клинитест.
Алкаптонурия
наследуется по аутосомнорецесспвному
типу. Гетерозиготных носителей не
удается обнаружить ни клиническими,
ни биохимическими методами.
Альбинизм.
Недостаточность тирознпазы в меланощггах
вызывает одну из форм альбинизма и
наследуется как рецессивный признак.
У пациентов отсутствует пигментация
кожи, волос и радужной оболочки
(глаза кажутся розовыми). Отсутствие
пигментации кожи и радужной оболочки
сопровождается острой фотосенсибилизацией.
Тирозиназа, участвующая в биосинтезе
катехоламинов, представляет собой
другой фермент, контролируемый иным
геном. У альбиносов метаболизм адреналина
в норме.
Нарушения
обмена других аминокислот
Болезнь
«кленового сиропа».
При болезни «кленового сиропа» происходит
недостаточное декарбоксилирование
оксокислот, образующихся в результате
дезаминирования трех аминокислот с
разветвленной цепью атомов углерода
в молекуле: лейцина, изолейцина и валина.
Эти аминокислоты накапливаются в крови
и экскретируются (вместе с соответствующими
оксокислотами) с мочой. В названии
болезни отражен тот факт, что запах
мочи больного напоминает запах
кленового сиропа.
Признаки
заболевания проявляются в течение
первой недели жизни. Если лечение не
проводится, то в течение нескольких
недель или месяцев развиваются тяжелые
поражения нервной системы и наступает
смерть. Но если заболевание диагностировано
и назначена диета, бедная аминокислотами
с разветвленной цепью атомов углерода,
то возможно нормальное развитие
ребенка.
Диагноз
ставится на основании повышенных
уровней аминокислот с разветвлено!!
цепью атомов углерода в крови и моче.
Дефицит соответствующих ферментов
в лейкоцитах может подтвердить
диагноз. Заболевание наследуется по
рецессивному типу.
Гистидинемия.
Гистидинемия связана с недостаточностью
фермента гистидазы, участвующего в
норме в метаболизме гистидина. При
данном патологическом состоянии в
крови повышена концентрация гистидина,
а в моче увеличено содержание гистидина
и его побочного метаболита
имидазолпировиноградной кислоты.
Подобно фенилиировиноградиои кислоте
(экскретируемой при фенилкетонурии),
имидазолпировиноградная кислота
реагирует с хлорным железом, образуя
синезеленый пигмент в присутствии
хлорного железа или препарата фенистикса.
Приблизительно в половине описанных
в литературе случаев отмечают умственную
отсталость, дефекты рсчп, у остальных
больных заметных отклонений от нормы
нет. Результаты диетотерапии, по
современным данным, не представляются
убедительными.
По-видимому,
заболевание наследуется по
аутосомнорецессивному типу.
Нарушения
обмена металлов
Два
врожденных патологических состояния
связаны с аномальным накоплением
металлов в организме. Чрезмерная
нагрузка железом (идиопатический
гемохроматоз) обсуждается в гл. XVIII.
Накопление меди происходит при болезни
Уилсона.
Болезнь
Уилсона (гепатолентикулярная дегенерация)
В
норме большая часть меди плазмы крови
входит в состав белка церулоплазмина,
но некоторое ее количество образует
непрочную связь с альбумином плазмы
крови. Экскреция меди происходит
преимущественно с желчью.
При
болезни Уилсона отмечают два нарушения
метаболизма меди: 1) нарушение экскреции
меди с желчью, что приводит к отложению
меди в печени; 2) недостаточность
церулоплазмина, результатом чего
является низкое содержание меди в
плазме крови. При этом медь преимущественно
находится в непрочно связанном виде;
ее отложение в тканях и фильтрация в
почечных клубочках происходят более
легко, чем в норме; экскреция меди с
мочой повышена.
Последствиями
чрезмерного отложения меди в базальных
ганглиях головного мозга, в печени,
почечных канальцах, тканях глаза
являются: 1) неврологические симптомы,
обусловленные дегенерацией базальных
ганглиев; 2) поражения печени, приводящие
к циррозу; 3) поражения почечных канальцев
с любыми (или всеми) биохимическими
проявлениями этого патологического
состояния, включая аминоацидурию
(синдром Фанкони); 4) кольца Кайзера —
Флейшера, окружающие роговую оболочку
вследствие отложения меди в десцеметовые
оболочки.
Диагностика.
У подавляющего большинства больных
концентрации церулоплазмина и меди
в плазме крови понижены.
Интерпретируя
результаты анализов содержания
церулоплазмина, важно помнить, что
низкая его концентрация возможна. также
при следующих обстоятельствах: 1) в
течение первых нескольких месяцев
жизни; 2) при недостаточности питания;
3) при нефротическом синдроме в связи
с потерями церулоплазмина с мочой.
Повышенные концентрации церулоплазмина
в крови могут быть в следующих
случаях: 1) при острых патологических
состояниях печени (что может объяснить
редко наблюдаемый нормальный уровень
церулоплазмина при болезни Уилсона);
2) в течение последнего триместра
беременности; 3) у женщин, принимающих
контрацептивные препараты; 4) при
неспецифических поражениях печени
(например, при воспалительных процессах
или новообразованиях).
В
некоторых случаях диагностическое
значение могут иметь результаты
определений ссодержания меди в образцах
печеночной ткани, полученных методом
биопсии.
Заболевание
наследуется по рецессивному типу, но
пониженные концентрации церулоплазмина
могут быть у гетерозиготных носителей.
Важно отличать их от гомозигот, у которых
клинических симптомов заболеваний
пока нет, хотя лечение следует проводить.
Цель
лечения с помощью таких образующих
внутрпкомплексные соединения с медью
веществ, как Dпеницилламин, — понижение
тканевой концентрации меди.
Лекарственные
средства и врожденные аномалии обмена
веществ
Вариации
индивидуальной чувствительности к
идентичным лекарственным средствам
отчасти могут быть предопределены
генетически. Существует целый ряд
хорошо изученных врожденных нарушений,
которые под воздействием определенных
лекарственных средств усугубляются
или даже выявляются клинически.
Подобные состояния можно разделить на
две группы.
Расстройства,
обусловленные нарушениями метаболизма
лекарств.
Миорелаксант дитилин в норме оказывает
весьма кратковременное действие,
так как он быстро распадается в результате
реакции, катализируемой холинэстеразой
плазмы крови. При повышенной
чувствительности к дитилину в организме
имеется аномальная холинэстераза,
распад лекарственного средства нарушен
и может наступить длительный респираторный
паралпч.
Два
других врожденных расстройства
характеризуются нарушением метаболизма
лекарственных средств изониазпда и
дифенина соответственно. При этих
расстройствах у больных отмечают
токсические явления чаще и под
воздействием более низких доз этих
препаратов, чем у здоровых.
Расстройства,
обусловленные аномальным ответом на
действие лекарств.
Одна из форм гемолитической анемии,
часто встречающаяся во многих регионах
мира, обусловлена недостаточностью
глюкозо6фосфатдегидрогеназы (Г6ФД) в
эритроцитах. Указанный фермент —
первый компонент гексозомонофосфатного
шунта, необходимый для образования
NADPH. В свою
очередь
NADPH, по-видимому,
необходим для поддержания целостности
биомембран эритроцитов. Описано
множество вариантов недостаточности
Г6ФД. Во многих случаях гемолиз усугубляли
лекарственные средства, в частности,
некоторые противомалярийные препараты,
такие как примахин. а также сульфонамиды
и аналоги витамина К.
При
врожденной печеночной порфирии острые
приступы могут быть спровоцированы
некоторыми лекарственными средствами,
в частности барбитуратами.
У
некоторых лиц препараты, применяемые
для общей анестезии (особенно часто
фторотан в сочетании с дитилином),
вызывают резкое повышение температуры
тела, ригидность мышц и ацидоз; при этом
большинство пациентов погибает
(злокачественная гиперпирексия).
Многие (но не все) члены семей этих
больных имеют повышенный уровень
креатинкиназы (КК).
Этот
краткий раздел должен напомнить
читателям о возможности врожденной
аномалии в тех случаях, когда имеется
несвойственная норме реакция
организма на воздействие лекарственного
средства. В конце данной главы мы
приводим ссылку на работу, характеризующую
эту развивающуюся область фармакогенетики.
ЗАКЛЮЧЕНИЕ
1.
Врожденные нарушения обмена веществ
могут вызывать заболевания, обусловленные
наследуемыми дефектами биосинтеза
белка. В большинстве случаев, когда
выражены клинические симптомы,
патологический процесс имеет в своей
основе нарушения биосинтеза белка.
2.
Врожденные нарушения обмена веществ
могут не сопровождаться развитием
клинических симптомов. Возможно
появление клинических симптомов только
при определенных обстоятельствах
(например, при наличии вариантов
холинэстеразы). С другой стороны,
врожденные аномалии метаболизма могут
вызывать тяжелые заболевания;
некоторые из них смертельны.
3.
Обнаружение определенных врожденных
аномалий обмена веществ представляет
только академический интерес. Диагностика
наследуемых аномалий метаболизма имеет
важное значение, если заболевание
является тяжелым, но поддающимся
лечению, если удается избежать воздействия
факторов, усугубляющих течение
заболевания, или если существуют
трудности при дифференцировании с
другими патологическими состояниями.
4.
Наследование заболеваний может быть
аутосомным или сцепленным с полом,
доминантным или рецессивным. По характеру
наследования патологические состояния,
приводящие к развитию тяжелых
клинических симптомов, преимущественно
бывают аутосомнорецессивными;
наиболее часто они встречаются в случаях
брака между кровными родственниками.
5.
Во многих случаях наследования клинически
диагностируемого заболевания по
рецессивному типу менее выраженные
аномалии можно обнаружить с помощью
биохимических тестов.
6.
В этой главе рассмотрены некоторые
врожденные нарушения метаболизма,
не упомянутые в других разделах данной
книги.
СПИСОК
ЛИТЕРАТУРЫ
Holton
J.
В.
Diagnosis of inherited metabolic diseases in severely ill children.—
Ann. Clin. Biochem.,
1982, 19, 389—395.
Holtzman
N.
A.
Newborn screening for inborn errors of metabolism.
—
Pediat, Clin.
N.
Amer.,
1978, 25. 411—
421
Seriver
С.
R., Clow
С.
L.
rheuvlkeloiniria: epitome of human biochemical genetics.
—
Now Engl. J. Med.,
I960, 303, 1336—1342
and
1394—140&.
Vesell
E. S.
Pharmacogenetics: multiple interactions between genes and
environment as determinants of drug response.
—
Amer. J. Med.,
1979, 66, 183— 187.
Stanbary
J. В.,
Wyngaarden J.
В.,
Fredrickson D. S., Goldstein J. L., Brown M. S.I Eds.
The metabolic basis of inherited disease, 5th edit., New York:
McGrawHill,
1983.
НЕКОТОРЫЕ
ВРОЖДЕННЫЕ НАРУШЕНИЯ ОБМЕНА И ИХ
НАСЛЕДУЕМОСТЬ
Приведенный
ниже перечень врожденных нарушений
метаболизма отнюдь не является
полным. Он предназначен только для
справок; не следует пытаться выучить
этот список. Большинство из перечисленных
аномалий мы обсуждаем в этой книге
(соответствующая страница указана). За
исключением тех случаев, когда
подзаголовок относится к группе
заболеваний с определенным типом
наследуемое™, мы указываем характер
наследования, если он известен.
D—аутосомнодоминантный; Р —
аутосомнорепессивный; Х-сцепленный;
D—Х-сцепленный доминантный; Х-сцепленный;
Р—Х-сцепленный рецессивный.
|
Наследуемость |
|
|
/. |
|
|
В |
|
|
Аминокислоты |
|
|
Двухосновные |
Р |
|
Нейтральные |
Р |
|
Наследственная |
Р |
|
Глюкоза |
|
|
Почечная |
D |
|
Вода |
|
|
Врожденный |
Х-сцепленный |
|
Натрий |
|
|
Псевдо-аддисонова |
? |
|
Калий |
|
|
Врожденный |
|
|
Кальций |
|
|
Псевдогипопаратиреоз |
Х-сцепленный |
|
Фосфаты |
|
|
Врожденная |
Х-сцепленный |
|
Ионы |
|
|
Почечно |
D |
|
Билирубин |
|
|
Врожденные |
|
|
Синдром |
P |
|
Синдром |
D |
|
Синдром |
?Р |
|
Болезнь |
? |
|
Синдром |
?Р |
|
//. |
|
|
Ароматические |
|
|
Фенилкетонурия |
Р |
|
Алкаптонурия |
Р |
|
Альбинизм |
Р |
|
Нарушения |
Все |
|
Серосодержащие |
|
|
Цистиноз |
Р |
|
Гомоцистинурия |
Р |
|
Аминокислоты |
|
|
Болезнь |
Р |
|
Гистидинемия |
Р |
|
//. |
|
|
Гликогенозы |
Р |
|
Галактоземия |
Р |
|
Врожденная |
Р |
|
Эссенциальная |
Р |
|
Сахарный |
? |
|
IV. |
|
|
Гиперлипопротеинемии |
— |
|
Гиполипопротеинемии |
Р |
|
Недостаточность |
Р |
|
V. |
|
|
Недостаточность |
— |
|
Аномалии |
|
|
Недостаточность |
?Р |
|
Недостаточность |
Х-сцепленный |
|
Вариации |
Р |
|
Недостаточность |
|
|
Бисальбуминемия |
D |
|
Анальбуминемия |
Р |
|
VI. |
|
|
Гемохроматоз |
? |
|
Болезнь |
Р |
|
VII. |
|
|
Гемоглобинопатии |
|
|
Недостаточность |
Х-сцепленный |
|
Недостаточность |
Р |
|
VIII. |
|
|
Порфирии |
— |
|
IX. |
|
|
Врожденные |
Все |
|
X. |
|
|
Первичная |
? |
|
Ксантинурия |
Р |
|
Синдром |
Х-сцепленный |
|
XI. |
|
|
Недостаточность |
Р |
|
Фиброкистоз |
Р |
|
XII. |
|
|
Первичная |
Р |
|
XIII. |
|
|
Чувствительность |
Р |
|
Медленная |
Р |
|
Токсичность |
D |
|
Недостаточность |
Х-сцепленный |
|
Злокачественная |
D |
В организме человека при участии специфических белков-ферментов протекают сотни различных биохимических реакций. Очевидно, что теоретически возможна мутация по любому структурному или регуляторному участку гена каждого фермента. В настоящее время известно более ста генных мутаций по определенным ферментным генам. По-видимому, некоторые из ферментных дефектов ведут к гибели особи и поэтому не обнаруживаются в популяции.
Фенилкетонурия является лишь одним из примеров врожденных ферментопатий. При этом заболевании ребенок развивается в психическом отношении замедленно, что приводит в более позднем возрасте к умственной отсталости. К заболеваниям, связанным с нарушением обмена аминокислот, относится также гистидинемия, при которой имеется врожденный дефект фермента гис-тидазы, расщепляющей гистидин. В организме накапливается в больших количествах гистидин и некоторые необычные продукты его распада. Клинические признаки гистидинемии — умственная отсталость, неразборчивая речь и сниженная пигментация кожи и волос.
Болезнь Менкеса названа так из-за характерного запаха мочи у новорожденных детей с этой патологией, напоминающего запах кленового сиропа. Врожденный дефект при этом редком заболевании состоит в невозможности нормального превращения в клетках аминокислот валина, лейцина и изолейцина. Состояние детей с болезнью кленового сиропа тяжелое, жизнеспособность снижена.
Встречается ряд наследственных ферментных дефектов, связанный с обменом аминокислот в цикле образования мочевины. При недостаточности каждого из пяти ферментов этого метаболического цикла мочевина накапливается в тканях и клетках и возникает тяжелое отравление организма, что сильнее всего отражается на функциях нервной системы.
Гомоцистинурия вызывается врожденным дефектом цистатионин-синтетазы — фермента, участвующего в образовании цистатионина. Это соединение, по-видимому, выполняет важные функции в нервной системе. Известно, что уровень цистатионина минимален у низших животных, значительно выше у обезьян и максимален у человека. Люди с гомоцистинурией имеют длинные конечности, костлявую фигуру. У них разболтаны суставы и ослаблены соединительнотканные связки скелета, весьма часты вывихи хрусталика глаза, что ведет к плохому зрению, имеется умственная отсталость.
Существуют также мутации генов ферментов углеводного обмена. На протяжении многих лет известны заболевания, именуемые гликогенозами, которые развиваются в результате генетических дефектов различных ферментов, участвующих в распаде гликогена, образовании и переработке глюкозы. Гликогенозы характеризуются различной клинической картиной и избыточным отложением гликогена в скелетной мускулатуре, сердечной мышце или печени. При некоторых формах гликогенозов могут развиться умственная отсталость, мышечная слабость или печеночная недостаточность.
Среди болезней углеводного обмена выделяется галактоземия, при которой имеется генетический дефект галактозо-1-фосфат уридилтрансферазы — фермента, переводящего молочный сахар (галактозу) в химически активированную форму. При этом заболевании в организме ребенка накапливается галактоза и ее фосфат. В клинической картине преобладают расстройства нервной системы с самого рождения, сильная желтуха. Возможна смерть ребенка. Диагноз основывается на результатах измерения активности данного фермента или обнаружении повышенных количеств галактозы в крови.
Генные мутации могут быть причиной ненормального обмена жиров и жироподобных веществ (липидов). Липидозы сопровождаются тяжелыми клиническими нарушениями — тяжелой умственной отсталостью и расстройством жизненно важных функций нервной системы. По-видимому, это объясняется особой значимостью нормального обмена липидов для нервной ткани.
Нередко встречаются наследственные дефекты обмена нуклеиновых кислот. Имеются основания думать, что большинство мутаций этих крайне важных генов приводят к смертельному исходу. Так, при заболевании оротовая ацидурия имеется блокада в синтезе пиримидиновых нуклеотидов. Подавляющее большинство мутантов, гомозиготных по гену оротовой ацидурии, скорее всего гибнет еще внутриутробно. Дети с этим заболеванием значительно умственно отсталы. В их органах и тканях определяются отложения оротовой кислоты.
Генетический дефект синтеза пуриновых оснований является причиной синдрома Леш — Найена, который передается сцепленно с полом, и поэтому болеют им только мальчики. При этом заболевании в почках, коже и других тканях накапливается мочевая кислота, может возникнуть подагра.
Клинически картина синдрома Леш — Найена весьма характерна. У больных развиваются параличи нижних конечностей, непроизвольные движения лицевых мышц и мускулатуры верхних конечностей. Характерна агрессивность, направленная на себя. Дети обгрызают себе губы и пальцы, наносят себе всякие повреждения.
Недавно стало известно о редкой генной мутации у человека, ведущей к повышению мутабельности генома больных. Специалисты по кожным болезням давно знают это состояние под названием «пигментная ксеродерма». У таких больных уже в первые годы жизни под влиянием ультрафиолетового облучения появляются тяжелые поражения кожных покровов и впоследствии развивается рак кожи. Работы генетиков показали, что при пигментной ксеродерме имеется мутация аутосомного гена. Вследствие этого в участках ДНК, повреждаемых ультрафиолетовым излучением, не происходит восстановления прежней, нормальной последовательности нуклеотидов. Тем самым мутации соматических клеток фиксируются. Мутантные клетки становятся маложизнеспособными или, наоборот, склонными к злокачественному росту.
В заключение расскажем о наиболее изученном классе генных мутаций человека. Исследования в 40-х годах XX века американского ученого Л. Полинга дополнили наши знания о гемоглобине — белке, переносящем кислород в организме. Гемоглобин имеет сложное строение. Его молекула состоит из глобина (четырех белковых цепей — две а- и две [5-цепи, различающихся по последовательности аминокислот) и молекулы гема (небелковое соединение). Человеческий плод в утробе матери содержит фетальный гемоглобин (так называемый гемоглобин Р), который функционирует наилучшим образом при тех биохимических условиях, какие имеются у плода. При рождении ребенка генетическая программа переключается на синтез гемоглобина типа А, характерного для взрослых.
Существует много различных генных мутаций гемоглобина, в основе которых лежит замена одной аминокислоты в глобиновой цепи на другую. Некоторые из них обнаруживаются только при массовых обследованиях населения.
Снижение способности гемоглобина к связыванию кислорода возникает лишь при некоторых мутациях. Так, в странах тропического пояса распространен так называемый гемоглобиноз. При этом заболевании гемоглобин мало растворим и выпадает в виде осадка внутри красных кровяных телец. Эритроциты приобретают вследствие этого характерную серповидную форму. У гомозиготных лиц развивается тяжелое малокровие с отложениями железа в различных органах и тканях. Имеются значительные нарушения нервной системы. Дети с гемоглобинозом маложизнеспособны. Интересно, что гетерозиготы по 5-гену обладают повышенной устойчивостью к малярии, распространенной в тропической местности.
Этим, по-видимому, и вызвана высокая частота данного гена у населения стран Африки и Южной Азии.
В различных районах мира, в частности в Средиземноморье, встречаются так называемые талассемии. При этих состояниях наблюдается врожденная недостаточность биосинтеза одной из глобиновых цепей. Так, р-талассемия характерна малой выработкой (3-цепей в клетках.
Таким образом, генные мутации у человека имеют громадное значение в медицинской практике. Их изучение может дать весьма ценные данные для понимания общебиологических механизмов и лечения наследственной патологии.