Groupthink is a collective set of systematic errors (biases) held by and perpetuated by a group.
From: Paradigms Lost, 2006
Experimental techniques
Yanqiu Huang, … Zhixiang Cao, in Industrial Ventilation Design Guidebook (Second Edition), 2021
4.3.3.2 Measurement errors
The measurement errors are divided into two categories: systematic errors and random errors (OIML, 1978).
Systematic error is an error which, in the course of a number of measurements carried out under the same conditions of a given value and quantity, either remains constant in absolute value and sign, or varies according to definite law with changing conditions.
Random error varies in an unpredictable manner in absolute value and in sign when a large number of measurements of the same value of a quantity are made under essentially identical conditions.
The origins of the above two errors are different in cause and nature. A simple example is when the mass of a weight is less than its nominal value, a systematic error occurs, which is constant in absolute value and sign. This is a pure systematic error. A ventilation-related example is when the instrument factor of a Pitot-static tube, which defines the relationship between the measured pressure difference and the velocity, is incorrect, a systematic error occurs. On the other hand, if a Pitot-static tube is positioned manually in a duct in such a way that the tube tip is randomly on either side of the intended measurement point, a random error occurs. This way, different phenomena create different types of error. The (total) error of measurement usually is a combination of the above two types.
The question may be asked, that is, what is the reason for dividing the errors into two categories? The answer is the totally different way of dealing with these different types. Systematic error can be eliminated to a sufficient degree, whereas random error cannot. The following section shows how to deal with these errors.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128166734000043
EXPERIMENTAL TECHNIQUES
KAI SIREN, … PETER V. NIELSEN, in Industrial Ventilation Design Guidebook, 2001
12.3.3.11 Systematic Errors
Systematic error, as stated above, can be eliminated—not totally, but usually to a sufficient degree. This elimination process is called “calibration.” Calibration is simply a procedure where the result of measurement recorded by an instrument is compared with the measurement result of a standard. A standard is a measuring device intended to define, to represent physically, to conserve, or to reproduce the unit of measurement in order to transmit it to other measuring instruments by comparison.1 There are several categories of standards, but, simplifying a little, a standard is an instrument with a very high accuracy and can for that reason be used as a reference for ordinary measuring instruments. The calibration itself is usually carried out by measuring the quantity over the whole range required and by defining either one correction factor for the whole range, for a constant systematic error, or a correction curve or equation for the whole range. Applying this correction to the measurement result eliminates, more or less, the systematic error and gives the corrected result of measurement.
A primary standard has the highest metrological quality in a given field. Hence, the primary standard is the most accurate way to measure or to reproduce the value of a quantity. Primary standards are usually complicated instruments, which are essentially laboratory instruments and unsuited for site measurement. They require skilled handling and can be expensive. For these reasons it is not practical to calibrate all ordinary meters against a primary standard. To utilize the solid metrological basis of the primary standard, a chain of secondary standards, reference standards, and working standards combine the primary standard and the ordinary instruments. The lower level standard in the chain is calibrated using the next higher level standard. This is called “traceability.” In all calibrations traceability along the chain should exist, up to the instrument with the highest reliability, the primary standard.
The question is often asked, How often should calibration be carried out? Is it sufficient to do it once, or should it be repeated? The answer to this question depends on the instrument type. A very simple instrument that is robust and stable may require calibrating only once during its lifetime. Some fundamental meters do not need calibration at all. A Pitot-static tube or a liquid U-tube manometer are examples of such simple instruments. On the other hand, complicated instruments with many components or sensitive components may need calibration at short intervals. Also fouling and wearing are reasons not only for maintenance but also calibration. Thus the proper calibration interval depends on the instrument itself and its use. The manufacturers recommendations as well as past experience are often the only guidelines.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780122896767500151
Intelligent control and protection in the Russian electric power system
Nikolai Voropai, … Daniil Panasetsky, in Application of Smart Grid Technologies, 2018
3.3.1.2 Systematic errors in PMU measurements
The systematic errors caused by the errors of the instrument transformers that exceed the class of their accuracy are constantly present in the measurements and can be identified by considering some successive snapshots of measurements. The TE linearized at the point of a true measurement, taking into account random and systematic errors, can be written as:
(25)wky¯=∑l∈ωk∂w∂ylξyl+cyl=∑aklξyl+∑aklcyl
where ∑ aklξyl—mathematical expectation of random errors of the TE, equal to zero; ∑ aklcyl—mathematical expectation of systematic error of the TE, ωk—a set of measurements contained in the kth TE.
The author of Ref. [28] suggests an algorithm for the identification of a systematic component of the measurement error on the basis of the current discrepancy of the TE. The algorithm rests on the fact that systematic errors of measurements do not change through a long time interval. In this case, condition (17) will not be met during such an interval of time. Based on the snapshots that arrive at time instants 0, 1, 2, …, t − 1, t…, the sliding average method is used to calculate the mathematical expectation of the TE discrepancy:
(26)Δwkt=1−αΔwkt−1+αwkt
where 0 ≤ α ≤ 1.
Fig. 5 shows the curve of the TE discrepancy (a thin dotted line) calculated by (26) for 100 snapshots of measurements that do not have systematic errors.

Fig. 5. Detection of a systematic error in the PMU measurements and identification of mathematical expectation of the test equation.
It virtually does not exceed the threshold dk = 0.014 (a light horizontal line). Above the threshold, there is a curve of the TE discrepancy (a bold dotted line) that contains a measurement with a systematic error and a curve of nonzero mathematical expectation Δwk(t) ∈ [0.026; 0.03] (a black-blue thick line). However, the nonzero value of the calculated mathematical expectation of the TE discrepancy can only testify to the presence of a systematic error in the PMU measurements contained in this TE, but cannot be used to locate it.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128031285000039
Measurements
Sankara Papavinasam, in Corrosion Control in the Oil and Gas Industry, 2014
ii Systematic or determinate error
To define systematic error, one needs to understand ‘accuracy’. Accuracy is a measure of the closeness of the data to its true or accepted value. Figure 12.3 illustrates accuracy schematically.4 Determining the accuracy of a measurement is difficult because the true value may never be known, so for this reason an accepted value is commonly used. Systematic error moves the mean or average value of a measurement from the true or accepted value.

FIGURE 12.3. Difference between Accuracy and Precision in a Measurement.4
Reproduced with permission from Brooks/Cole, A Division of Cengage Learning.
Systematic error may be expressed as absolute error or relative error:
- •
-
The absolute error (EA) is a measure of the difference between the measured value (xi) and true or accepted value (xt) (Eqn. 12.5):
(Eqn. 12.5)EA=xi−xt
Absolute error bears a sign:
- •
-
A negative sign indicates that the measured value is smaller than true value and
- •
-
A positive sign indicates that the measured value is higher than true value
The relative error (ER) is the ratio of measured value to true value and it is expressed as (Eqn. 12.6):
(Eqn. 12.6)ER=(xi−xtxt).100
Table 12.2 illustrates the absolute and relative errors for six measurements in determining the concentration of 20 ppm of an ionic species in solution.
Table 12.2. Relative and Absolute Errors in Six Measurements of Aqueous Solution Containing 20 ppm of an Ionic Species
| Measured Value | Absolute Error | Relative Error (Percentage) | Remarks |
|---|---|---|---|
| 19.4 | −0.6 | −3.0 | Experimental value lower than actual value. |
| 19.5 | −0.5 | −2.5 | |
| 19.6 | −0.4 | −2.0 | |
| 19.8 | −0.2 | −1.0 | |
| 20.1 | +0.1 | +0.5 | Experimental value higher than actual value. |
| 20.3 | +0.3 | +1.5 |
Systematic error may occur due to instrument, methodology, and personal error.
Instrument error
Instrument error occurs due to variations that can affect the functionality of the instrument. Some common causes include temperature change, voltage fluctuation, variations in resistance, distortion of the container, error from original calibration, and contamination. Most instrument errors can be detected and corrected by frequently calibrating the instrument using a standard reference material. Standard reference materials may occur in different forms including minerals, gas mixtures, hydrocarbon mixtures, polymers, solutions of known concentration of chemicals, weight, and volume. The standard reference materials may be prepared in the laboratory or may be obtained from standard-making organizations (e.g., ASTM standard reference materials), government agencies (e.g., National Institute of Standards and Technology (NIST) provides about 900 reference materials) and commercial suppliers. If standard materials are not available, a blank test may be performed using a solution without the sample. The value from this test may be used to correct the results from the actual sample. However this methodology may not be applicable for correcting instrumental error in all situations.
Methodology error
Methodology error occurs due to the non-ideal physical or chemical behavior of the method. Some common causes include variation of chemical reaction and its rate, incompleteness of the reaction between analyte and the sensing element due to the presence of other interfering substances, non-specificity of the method, side reactions, and decomposition of the reactant due to the measurement process. Methodology error is often difficult to detect and correct, and is therefore the most serious of the three types of systematic error. Therefore a suitable method free from methodology error should be established for routine analysis.
Personal error
Personal error occurs due to carelessness, lack of detailed knowledge of the measurement, limitation (e.g., color blindness of a person performing color-change titration), judgment, and prejudice of person performing the measurement. Some of these can be overcome by automation, proper training, and making sure that the person overcomes any bias to preserve the integrity of the measurement.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123970220000121
Experimental Design and Sample Size Calculations
Andrew P. King, Robert J. Eckersley, in Statistics for Biomedical Engineers and Scientists, 2019
9.4.2 Blinding
Systematic errors can arise because either the participants or the researchers have particular knowledge about the experiment. Probably the best known example is the placebo effect, in which patients’ symptoms can improve simply because they believe that they have received some treatment even though, in reality, they have been given a treatment of no therapeutic value (e.g. a sugar pill). What is less well known, but nevertheless well established, is that the behavior of researchers can alter in a similar way. For example, a researcher who knows that a participant has received a specific treatment may monitor the participant much more carefully than a participant who he/she knows has received no treatment. Blinding is a method to reduce the chance of these effects causing a bias. There are three levels of blinding:
- 1.
-
Single-blind. The participant does not know if he/she is a member of the treatment or control group. This normally requires the control group to receive a placebo. Single-blinding can be easy to achieve in some types of experiments, for example, in drug trials the control group could receive sugar pills. However, it can be more difficult for other types of treatment. For example, in surgery there are ethical issues involved in patients having a placebo (or sham) operation.2
- 2.
-
Double-blind. Neither the participant nor the researcher who delivers the treatment knows whether the participant is in the treatment or control group.
- 3.
-
Triple-blind. Neither the participant, the researcher who delivers the treatment, nor the researcher who measures the response knows whether the participant is in the treatment or control group.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780081029398000189
The pursuit and definition of accuracy
Anthony J. Martyr, David R. Rogers, in Engine Testing (Fifth Edition), 2021
Systematic instrument errors
Typical systematic errors (Fig. 19.2C) include the following:
- 1.
-
Zero errors—the instrument does not read zero when the value of the quantity observed is zero.
- 2.
-
Scaling errors—the instrument reads systematically high or low.
- 3.
-
Nonlinearity—the relation between the true value of the quantity and the indicated value is not exactly in proportion; if the proportion of error is plotted against each measurement over full scale, the graph is nonlinear.
- 4.
-
Dimensional errors—for example, the effective length of a dynamometer torque arm may not be precisely correct.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B978012821226400019X
Power spectrum and filtering
Andreas Skiadopoulos, Nick Stergiou, in Biomechanics and Gait Analysis, 2020
5.10 Practical implementation
As suggested by Winter (2009), to cancel the phase shift of the output signal relative to the input that is introduced by the second-order filter, the once-filtered data has to filtered again, but this time in the reverse direction. However, at every pass the −3dB cutoff frequency is pushed lower, and a correction is needed to match the original single-pass filter. This correction should be applied once the coefficients of the fourth-order low-pass filter are calculated. Nevertheless, it should be also checked whether functions of closed source software use the correction factor. If they have not used it, the output of the analyzed signal will be distorted. The format of the recursive second-order filter is given by Eq. (5.36) (Winter, 2009):
(5.36)yk=α0χk+α1χk−1+α2χk−2+β1yk−1+β2yk−2
where y are the filtered output data, x are past inputs, and k the kth sample.The coefficients α0,α1,α2,β1, and β2 for a second-order Butterworth low-pass filter are computed from Eq. (5.37) (Winter, 2009):
(5.37)ωc=tanπfcfsCK1=2ωcK2=ωc2K3=α1K2α0=K21+K1+K2α1=2α0α2=α0β1=K3−α1β2=1−α1-K3
where, ωc is the cutoff angular frequency in rad/s, fc is the cutoff frequency in Hz, and fs is the sampling rate in Hz. When the filtered data are filtered again in the reverse direction to cancel phase-shift, the following correction factor to compensate for the introduced error should be used:
(5.38)C=(21n−1)14
where n≥2 is the number of passes. For a single-pass C=1, and no compensation is needed. For a dual pass, (n=2), a compensation is needed, and the correction factor should be applied. Thus, the ωc term from Eq. (5.37) is calculated as follows:
(5.39)ωc=tan(πfcfs)(212−1)14=tan(πfcfs)0.802
A systematic error is introduced to the signal if the correction factor is not applied. Therefore, remember to check any algorithm before using it. Let us check the correctness of the fourth-order low-pass filter that was built previously in R language. Vignette 5.2 contains the code to perform Winter’s (2009) low-pass filter in R programming language. Because the filter needs two past inputs (two data points) to compute a present filtered output (one data point), the time-series data to be filtered (the raw data) should be padded at the beginning and at the end. Additional data are usually collected before and after the period of interest.
Vignette 5.2
The following vignette contains a code in R programming language that performs the fourth-order zero-phase-shift low-pass filter from Eq. (5.37).

- 1.
-
The first step is to create a sine (or equally a cosine) wave with known amplitude and known frequency. Vignette 5.3 is used to synthesize periodic digital waves. Let us create a simple periodic sine wave s[n] with the following characteristics:
- a.
-
Amplitude A=1 unit (e.g., 1 m);
- b.
-
Frequency f=2 Hz;
- c.
-
Phase θ=0 rad;
- d.
-
Shift a0=0 unit (e.g., 0 m).
Vignette 5.3
The following vignette contains a code in R programming language that synthesizes periodic waveforms from sinusoids.

Let us choose an arbitrary fundamental period T0=2 seconds, which corresponds to a fundamental frequency of f0=1/T0=0.5 Hz. Now, knowing the fundamental frequency, the fourth harmonic that corresponds to a sine wave with frequency of f=2 Hz will be chosen. The periodic sine wave s[n] will be sampled at Fs=40 Hz (Ts=1/40 seconds) (i.e., 20 times the Nyquist frequency, fN=2 Hz). The sine wave will be recorded for a time interval of t=2 s, which corresponds to N=80 data points. Thus, and because ω0=2πf0, we have:
s[n]=sin(2ω0nTs)
which means that the fourth harmonic has frequency f=2
Hz. Fig. 5.14A shows the sine wave created. The first and last 20 data points can be considered as extra points (padded). Additional data at the beginning and end of the signal are needed for the next steps because the filter is does not behave well at the edges. Thus, the signal of interest starts at 0.5
seconds and ends at 1.5
seconds, which corresponds to N=40 data points.

Figure 5.14. (A) Example of a low-pass filter (cutoff frequency=2 Hz) applied to a sine wave sampled at 40 Hz, with amplitude equal to 1 m, and frequency equal to 2 Hz. (B) The signal interpolated by a factor of 2, and filtered with cutoff frequency equal to the frequency of the sine wave (cutoff frequency=2 Hz). (C) Since the amplitude of the filtered signal has been reduced by a ratio of 0.707, the low-pass filter correctly attenuated the signal. The power spectra of the original and reconstructed signal are shown.
- 2.
-
An extra, but not mandatory, step is to interpolate the created sine wave in order to increase the temporal resolution of the created signal (Fig. 5.14B). Of course, when a digital periodic signal is created from scratch, like we are doing using the R code in the vignettes, we can easily sample the signal at higher frequencies. However, if we want to use real biomechanical time series data, that have already been collected, a possible way to increase its temporal resolution is by using the Whittaker–Shannon interpolation formula. With the Whittaker–Shannon interpolation a signal is up-sampled with interpolation using the sinc() function (Yaroslavsky, 1997):
(5.40)s(x)=∑n=0N−1αnsin(π(xΔx−n))Nsin(π(xΔx−n)/N)
The Whittaker–Shannon interpolation formula can be used to increase the temporal resolution after removing the “white” noise from the data. Without filtering, the interpolation results in a noise level equal to that of the original signal before sampling (Marks, 1991). However, the interpolation noise can be reduced by both oversampling and filtering the data before interpolation (Marks, 1991). An alternative, and efficient, method is to run the DFT, zero-pad the signal, and then take the IDFT to reconstruct it. Vignette 5.4 can be used to increase the temporal resolution by a factor of 2, which corresponds to a sampling frequency of 80 Hz.
Vignette 5.4
The following vignette contains a code in R programming language that runs the normalized discrete sinc() function, and the Whittaker–Shannon interpolation function.

- 3.
-
The third step is to filter the previously created sine wave with the fourth-order zero-phase-shift low-pass filter, setting the cutoff frequency equal to the sine wave frequency f=2 Hz. Vignette 5.2 is used for step 3. To cancel any shift (i.e., a zero-phase-shift filter) n=2 passes must be chosen. The interpolated signal has a sampling rate of 80 Hz.
- 4.
-
The fourth step is to investigate the frequency response of the filtered sine wave. The frequency response of the Butterworth filter is given by Eq. (5.41)
(5.41)|AoutAin|=1(1+fsf3dB)2n
where the point at which the amplitude response, Aout, of the input signal, s[n], with frequency, f, and amplitude, Ain, drops off by 3dB and is known as the cutoff frequency, f3dB. When the cutoff frequency is set equal to the frequency of the signal (f3dB=f), the ratio should be equal to 0.707, since:
(5.42)|AoutAin|=12≈0.707
Fig. 5.14C shows the plots of the filtered and interpolated sine wave. Since the ratio of the maximum value of the filtered sine wave to the original sine wave ratio=0.707, the created fourth-order zero-phase-shift low-pass filter works properly. Without the correction factor the amplitude reduces nearly to half (0.51), indicating that the coefficients of the filter needed correction. Fig. 5.15 also shows an erroneously filtered signal. You can try to create Fig. 5.15 by yourself.

Figure 5.15. Example of a recursive low-pass filter applied to a sine wave with amplitude equal to 1 m and cutoff frequency equal to the frequency of the sine wave. Since the amplitude of the filtered signal has been reduced by a ratio of 0.707, the low-pass filter correctly attenuated the signal. However, the function without the correction factor reduced the amplitude by nearly one-half (0.51), indicating that the coefficients need correction.




The same procedure should be applied to check whether the output of the functions from closed source software used the correction factor or not. For example, using the library(signal) of the R computational software, if x is the vector that contains the raw data, then using butter() the Butterworth coefficients can be generated.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128133729000051
Introduction to coal sampling
Wes B. Membrey, in The Coal Handbook (Second Edition), 2023
4.1.1 Definitions
The following definitions have been adapted from definitions given in the sampling standards.
Accuracy. A measure of the closeness of agreement between an analytical result and the true value or a reference value.
Cut. An increment taken by a sampling device typically from a conveyor belt, screen discharge, or other streams of coal.
Bias. Systematic error which leads to the average value of a series of analytical results being persistently higher or lower than the true value or a reference sample result.
Error. Difference between the measured value and the true value or a value from a reference sample result.
Increment. An amount of coal taken from a body of coal (a truck or barge, etc.) or from a stream of coal (coal on a conveyor, sizing screen or a chute, etc.) in a single operation of the sampling device.
Lot. Defined quantity of coal for which the quality is to be determined.
Particle top size is the nominal top size and is the square aperture size of the smallest sieve through which 95% of the sample passes.
Precision. A statistical term that quantifies how closely repeated experimental values agree. It usually has the value of the 95% confidence level, or 2 standard deviations from the mean of the experimental values.
Representative. A sample is representative when the sampling error, a combination of precision and accuracy, is of an acceptable level.
Sample. Quantity of coal with qualities that are representative of a larger mass (lot) for which the quality is to be determined.
Standard deviation. A measure of the spread of a set of values, equal to the square root of the variance of the results.
Sub lot. A part of a lot that is sampled and tested separately to the entire lot.
Tolerance. The maximum acceptable difference between measurement values or analytical results.
Variance. A measure of the spread of a set of values expressed as the square of the differences between the values and their mean.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128243282000169
Sensors
Andrea Colagrossi, … Matteo Battilana, in Modern Spacecraft Guidance, Navigation, and Control, 2023
Quantization errors
Quantization error is a systematic error resulting from the difference between the continuous input value and its quantized output, and it is like round-off and truncation errors. This error is intrinsically associated with the AD conversion that maps the input values from a continuous set to the output values in a countable set, often with a finite number of elements. The quantization error is linked to the resolution of the sensor. Namely, a high-resolution sensor has a small quantization error. Indeed, the maximum quantization error is smaller than the resolution interval of the output, which is associated to the least significant bit representing the smallest variation that can be represented digitally:
LSB=FSR2NBIT
where FSR is the full-scale range of the sensor, and NBIT is the number of bits (i.e., the resolution) used in the AD converter to represent the sensor’s output. Quantization errors are typically not corrected, and the discrete values of the output are directly elaborated by the GNC system, which is designed to operate on digital values.
Fig. 6.9 shows a convenient model block to simulate quantization errors.

Figure 6.9. Quantization error model.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323909167000068
The Systems Approach to Control and Instrumentation
William B. Ribbens, in Understanding Automotive Electronics (Seventh Edition), 2013
Systematic Errors
One example of a systematic error is known as loading errors, which are due to the energy extracted by an instrument when making a measurement. Whenever the energy extracted from a system under measurement is not negligible, the extracted energy causes a change in the quantity being measured. Wherever possible, an instrument is designed to minimize such loading effects. The idea of loading error can be illustrated by the simple example of an electrical measurement, as illustrated in Figure 1.17. A voltmeter M having resistance Rm measures the voltage across resistance R. The correct voltage (vc) is given by

Figure 1.17. Illustration of loading error-volt meter.
(71)vc=V(RR+R1)
However, the measured voltage vm is given by
(72)vm=V(RpRp+R1)
where Rp is the parallel combination of R and Rm:
(73)Rp=RRmR+Rm
Loading is minimized by increasing the meter resistance Rm to the largest possible value. For conditions where Rm approaches infinite resistance, Rp approaches resistance R and vm approaches the correct voltage. Loading is similarly minimized in measurement of any quantity by minimizing extracted energy. Normally, loading is negligible in modern instrumentation.
Another significant systematic error source is the dynamic response of the instrument. Any instrument has a limited response rate to very rapidly changing input, as illustrated in Figure 1.18. In this illustration, an input quantity to the instrument changes abruptly at some time. The instrument begins responding, but cannot instantaneously change and produce the new value. After a transient period, the indicated value approaches the correct reading (presuming correct instrument calibration). The dynamic response of an instrument to rapidly changing input quantity varies inversely with its bandwidth as explained earlier in this chapter.

Figure 1.18. Illustration of instrument dynamic response error.
In many automotive instrumentation applications, the bandwidth is purposely reduced to avoid rapid fluctuations in readings. For example, the type of sensor used for fuel-quantity measurements actually measures the height of fuel in the tank with a small float. As the car moves, the fuel sloshes in the tank, causing the sensor reading to fluctuate randomly about its mean value. The signal processing associated with this sensor is actually a low-pass filter such as is explained later in this chapter and has an extremely low bandwidth so that only the average reading of the fuel quantity is displayed, thereby eliminating the undesirable fluctuations in fuel quantity measurements that would occur if the bandwidth were not restricted.
The reliability of an instrumentation system refers to its ability to perform its designed function accurately and continuously whenever required, under unfavorable conditions, and for a reasonable amount of time. Reliability must be designed into the system by using adequate design margins and quality components that operate both over the desired temperature range and under the applicable environmental conditions.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780080970974000011
1) Кислотно-основного титриметрического определения уксусной кислоты в уксусной эссенции;
2) Гравиметрического определения хроматов в электролите для хромирования.
Абсолютная погрешность аналитических
весов 0,1мг
Абсолютная погрешность (ошибка)
∆x=xi—xист.
Xi-измеренное
значениеXист-истинное
значение ( если истинное значение не
известно – берется среднее)
Абсолютная погрешность не может ясно
охарактеризовать точность измерения,
так как она не связана с измеренным
значением.
Относительная погрешность (ошибка)
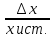 ·100%
·100%
Систематические погрешности (ошибки)– возникают при действии постоянных
причин, их можно выявить устранить или
учесть изменяются по постоянно
действующему закону .
-
Инструментальные погрешности–связанные с инструментами для измерения
аналитического сигнала (весы, посуда)
уменьшить можно периодической проверкой
аналитических приборов. Обычно составляют
небольшую долю . -
Методические ошибки–
обусловлены методом анализа (например
погрешности пробоотбора и пробоподготовки.)
вносят основной вклад в общую погрешность. -
Реактивные– связаны с чистотой
используемых реактивов. -
Оперативные ошибки –зависят
от правильности и точности выполнения
аналитических операций (например,
недостаточное или излишнее промывание
или прокаливания осадков, недостаточное
тщательное перемещение осадка из одной
посуды в другую, неправильный способ
выливания раствора из пипетки и т.д.) -
Индивидуальные ошибки(личные) – это результат некоторых
физических недостатков экспериментатора,
которые мешают ему правильно проводить
известные операции.
Способы выявления систематических
погрешностей
1)варьирование величин пробы
Увеличив размер в кратное число раз
можно обнаружить по изменению найденного
содержания постоянную систематическую
погрешность
2)способ «введено найдено»
Добавить точно известное количество
компонента в той же форме, в которой
находится аналитический объект. Введенная
добавка проводится через все стадии
анализа. Если на конечной стадии
определяется добавка с точностью, то
систематической ошибки нет.
3) сравнение результата анализа с
результатом, полученным другим независимым
методом
4)анализ стандартного образца
Проведение всех стадий анализа, на
стадии обработки сравнивается с
паспортом, если все совпадает , то
систематической ошибки нет.
Типы погрешностей
-
Погрешности известной природы, могут
быть рассчитаны и учтены введение
соответствующей поправки -
Погрешности известной природы, значение
которых может быть оценены в ходе
химического анализа
Релятивизация — способ устранения
систематической погрешности, когда в
идентичных условиях проводят отдельные
аналитические операции таким образом,
что происходит нивелирование
систематической ошибки
-
Погрешность невыясненной природы,
значение который неизвестно, их сложно
выявить и устранить , используют прием
рандомизации
Рандомизация – переведение систематической
ошибки в разряд случайной
Случайные ошибки– обрабатываются
по правилам матемтической статистики,
связаны с влиянием неконтролируемых
параметров, непредвиденны и неучтимы.
Промахи– грубые ошибки, сильно
искажающие результаты анализа (ошибки
при расчётах, неправильный отчёт по
шкале, проливание раствора или просыпание
осадка). Результат с промахом отбрасывается
при выводе среднего значения.
6. Случайные
ошибки. Метрологические характеристики,
отражающие случайные ошибки. Оценка и
критерии воспроизводимости и правильности.
Рассмотрите на примере титриметрического
комплексонометрического определения
меди (II).
Случайные ошибки–отражают
неопределенность результата , присущую
любому измерению, обрабатываются по
правилам матемтической статистики,
связаны с влиянием неконтролируемых
параметров, непредвиденны и неучтимы.
Причины таких погрешностей:
Изменение температуры во время измерения,
ослабление внимания при работе, случайные
потери, загрязнение, использование
разной посуды, весов и тд.
метрологические характеристики:
Правильность— характеризует степень
близости измеренного результата
некоторой величины к её истинному
значению
Воспроизводимость— характеризует
степень близости друг к другу единичный
определений (рассеяние единичных
результатов относительно среднего
значения
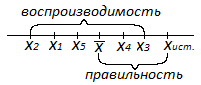
Точность— собирательная характеристика
метода или методики , включающая их
правильность и воспроизводимость .
Чувствительность— величина,
определяемая минимальным количеством
вещества, которое можно обнаружить
данным методом
Чувствительность – собирательное
понятие , включающее три характеристики:
1)Коэффициент чувствительности
коэффициент чувствительности sхарактеризует отклик аналитического
сигналаyна содержание
компонентаc,s-
это значение первой производной
градуировочной функции при определенном
содержании компонента, для прямолинейных
градуировочных графиковs– это тангенс угла наклона прямойy=Sc+b
s=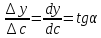
чем больше s, тем меньшие
количества компонента можно обнаружить
, используя один и тот же аналитический
сигнал, чем большеs, тем
точнее можно определить одно и то же
количество вещества
2)предел обнаружения Сminнаименьшее содержание при котором по
данной методике можно обнаружить
присутствие компонента с заданной
доверительной вероятностью, относится
к области качественного анализа и
определяет минимальное содержание
компонента
3)нижняя граница определяемого содержания
Сн
В количественном анализе обычно приводят
интервал определяемых содержаний-
область значений определяемых содержаний,
предусмотренная данной методикой и
ограниченная нижней и верхней границами.
Верхняя граница Свнаибольшее
значение количества или концентрации
компонента, определяемое по данной
методике.
нижняя граница Сн-наименьшее
содержание компонента , определяемое
по данной методике . З нижнюю границу
обычно принимают то минимальное
количество или концентрацию, которые
можно определить с относительным
стандартным отклонением Ϭr≤0,33
Оценка и критерии воспроизводимости
1)Среднее арифметическое
 =
=
2)Отклонение
di=xi—
3)Медиана— тот единичный результат
, относительно которого число результатов
с большими и меньшими значениями
одинаковое, если количество значений
нечетное, то медиана совпадает с
центральным результатом ранжированной
выборки , если количество значений
четное, то медиана есть среднее
арифметическое между двумя центральными
значениями ранжированной выборки
4)среднее отклонение-среднее
арифметическое единичных отклонений,
без учет знака
 =
=
5)Дисперсия
Ϭ2илиs2
Ϭ2=
еслиn>10
Ϭ2=
еслиn≤10
6)стандартное отклонениеϬx=
7)Относительное стандартное отклонение
Ϭr=
Титриметрическое комплексонометрическое
определения меди (II).
Выполнение определениея
1)Титрование исследуемого раствора
стандартным раствором ЭДТА
2)расчет граммового содержания меди
Ход анализа:Титрование исследуемого
раствора стандартным раствором ЭДТА.
Анализируемый раствор помещают в мерную
колбу на 100 мл, довдят водой до метки,
тщательно перемешивают. В коническую
колбу дл титрования берут аликвоту,
добавляют индикатор мурексид на кончике
шпателя и титруют раствором ЭДТА сначала
до грязно-розового цвета, натем добавляют
несколько капель 10%-ного раствора аммиака
до появления изумрудной или желтой
окраски раствора и дотитровывают
раствором ЭДТА до перехода окраски в
фиолетовую.
Формула для расчета граммового содержания
меди:
mCu,г=C( ЭДТА)·
ЭДТА)· ЭДТА·K
ЭДТА·K
ЭДТА·Mэкв(Cu)·P·10-3
Формула для расчета процентного
содержания меди:
ωCu= ·100%
·100%
Возможные причины возникновения
случайных ошибокв комплексонометрическом
титровании меди возникают в процессе
измерения объемов: неточное доведение
до метки мерной колбы, использование
разных пипеток, потеря титранта (капнуло
мимо), использование непромытой посуды.
Так же могут возникать ошибки из-за
неточного определения перехода окраски
, но эти ошибки будут относиться к
категории систематических индивидуальных
ошибок.
7. Гравиметрическое
определение бария в минерале альстонит:
этапы определения, возможные формулы
осадителей, осаждаемой и гравиметрической
формы, механизм образования осадка,
возможные варианты загрязнения осадка,
приемы повышения чистоты осадка,
погрешности определения. Условия
аналитического выделения осадков бария.
Минерал альстонит минерал, безводный
двойной карбонат бария и кальция
BaCa(CO3)2
Этапы определения:
1)взятие навески и её растворение
2)расчет количества осадителя
3)приготовление раствора осадителя
4)осаждение
5)фильтрование и промывание
6)высушивание и прокаливание осадка
7)взвешивание осадка, расчёт содержания
бария
Для количественного определения бария
его осаждают в виде сульфата BaSO4
(осаждаемая форма)
BaCO3+H2SO4=
BaSO4+H2CO3
В качестве осадителя, посташика
сульфат-ионов используют серную кислоту
H2SO4(осадитель)
После прокаливания осадка его формула
не меняется и остается так же в виде
сульфата бария BaSO4
(гравиметрическая форма)
Механизм образования осадка:
В процессе образования осадка различают
три стадии :
1)образование зародышей кристаллов
2)рост кристаллов
3)объединение (агрегация) хаотично
ориентированных кристаллов
Насыщение=>пересыщение=>ПКИ>ПР=>
образование мельчайших зародышей
кристаллов
Осаждение происходит при определенной
степени пересыщения раствора
P= =
= s-растворимость,
s-растворимость, -относительное
-относительное
пересыщение,Q-концентрация
кристаллизующегося вещества в растворе
Центром кристалла может служить твердая
частица этого вещества или любая другая
твердая частица, которую мы вносим в
раствор, твердые частицы могут изначально
присутствовать в растворе как примесь.
Если осаждение происходи из разбавленных
растворов, то появление осадка занимает
время-индукцинный период.
В процессе добавления каждой новой
порции осадителя происходит мгновенное
пересыщение раствора, зародыши растут
быстро за счет окружающих их ионов, как
только зародыш дотиг определенного
размера выпадает осадок
Рост кристаллов идет параллельно 1-ой
стадии, происходит за счет диффузии
ионов к поверхности растущего кристалла.
Число и размер частиц осадка (дисперсность
системы кол-во в единицы объёма) зависит
от соотношения скоростей 1-ой и 2-ой
стадий
V1— скорость образования
зародышейV2-скорость
роста кристаллов
V1>>V2-мелкодисперсный
осадокV1<<V2-крупнокристаллический
осадок
Лимитирующую стадию определяет скорость
осаждения и концентрации ионов
При медленном осаждении лимитирующей
стадией является кристаллизация ,
частица окружена однородным слоем
осаждаемый ионов в результате получается
кристалл правильной форм
При высокой концентрации ионов
лимитирующей стадией становится диффузия
, образуются кристаллы не правильной
формы с большой площадью поверхности
Следует отметить, что на скорость
процесса кристаллизации влияет
 ,
,
влияние различно на скорость образования
различно на скорость образования
зародышей и на скорость роста кристаллов
В случае образования зародышей
V1=k·( экспоненциальный
экспоненциальный
закон
В случае роста кристаллов V2=k·
При высокой степени
 образуются
образуются
мелкодисперсные осадки, при уменьшении ,
,
образуются крупнокристаллические
осадки
Агрегация происходит в гетерогенной
системе, в значительной степени
определяется числом центров кристаллизации.
Чем больше центров кристаллизации , тем
в меньшей степени они укрупняются на
второй стадии , тем хуже структура и тем
выше дисперсность осадков.
К аналитическим свойствам осадка
относятся: растворимость, чистота,
фильтруемость.
Лучшими свойствами обладают
крупнокристаллические осадки.
Загрязнение осадков
В гарвиметрическом определении часто
возникают ошибки , вызванные переходом
осадка в раствор или веществ из раствора
в осадок-соосождение
Соосаждение происходит в процессе
образования осадка
Отрицательная роль : загрязнение осадка
Положительная роль :используется для
концентрирования микропримесей
Существует три типа соосаждения:
1)Адсорбция- соосаждение примесей на
поверхности уже сформированного осадка,
происходит в результате нескомпенсированности
зарядов внутри и на поверхности.
Характеризуется ярко выраженной
избирательность, преимущественно
адсорбируются те ионы, которые входят
в структуру осадка, противоионы-примеси
Адсорбция противоионов подчиняется
правилам Панета-Фаянса-Гана
А)при одинаковых концентрациях
адсорбируются многозарядные ионы
Б)при одинаковых зарядах адсорбируются
те, концентрация которых выше
В)при одинаковых концентрациях и
зарядах-те, которые образуют с ионами
решетки менее растворимое соединение
Г)в кислой среде соосаждение ионов
уменьшается в следствии конкурентной
адсорбции H3O+
Количество адсорбируемой примеси
зависит от величины поверхности осадка,
концентрации адсорбируемой примеси и
температуры ( с ↑ поверхности и ↑
концентрации- адсорбция ↑; с ↑ температуры
адсорбция ↓)
2)Окклюзия- загрязнение осадка в результате
захвата примеси внутрь растущего
кристалла, происходит в процессе
формирования осадка.
Различают 2-х видов: абсорбционная и
механическая
Механическа- случайный захват частиц
маточного раствора внутрь твердой фазы
вследствие нарушения механической
структуры
Характерна при выделении аморфных
осадков.
Окклюзированные примеси равномерно
распределены внутри, но не принимают
участие в построении решетки кристалла.
Адсорбционная-возникает при быстром
росте кристалла, когда ионы на поверхности
обратают кристаллизованным веществом.
Протекает вследствии адсорбции примесей
по микротрещинам кристаллической
структуры.
Окклюзия подчиняется тем же правилам,
что и адсорбция
Общие правила понижения окклюзии–замедление процесса выделения твердой
фазы-осаждение при малом пересыщении
, работают с разбавленными растворами
, осадитель добавляют по каплям, при
постоянном перемешивании.
3)изоморфное соосаждение характерно
для изоморфно кристаллизующегося
веществ, которые могут образовывать
смешанные кристаллы, примесь участвует
в построении кристаллической решетки,
наблюдается лишь в тех случаях, когда
вещества сходны по химическим свойствам
или ионы имеют одинаковые кч и радиус.
Совместное осаждение-выделение в твердую
фазу нескольких веществ, для которых в
услових осаждения достигнуты величины
их Kst
Последовательное осаждение- веделение
примеси на поверхности уже сформированного
осадка
Приемы и методы повышения чистоты
осадка
Зависят от типа соосаждения
1)адсорбционные примеси хорошо удаляются
промыванием осадка, более эффективно
многократное промывание малыми порциями
Выбор промывочной жидкости:
Не увеличивает растворимость осадка и
не ухудшает его фильтруемость, водой
промывают осадки с k~10-11/-12,
не подвергаемых пептизации, кристаллические
осадки с конст, растворимости 10-9/-11промывают разбавленным раствором
осадителя, аморфные осадки промывают
разбавленными растворами электролитов
коагуляторов, чтобы избежать пептизации
Промывние кристаллических осадков
проводят холодной промывочной жидкостью,
чтоб не увеличивать растворимость,
аморфные наоборот горячими
2)окклюзированные примеси , для избавления
от них:
Для кристаллических осадков-старение
Для аморфных-переосаждение
Погрешность гравиметрического
метода анализа
Общая погрешность анализа
Ϭ2=
 +
+
 -погрешность
-погрешность
пробоотбораm-число пробn-число параллельных
определений
 -погрешность
-погрешность
измерений
Результат находится по формуле
P,%= ·100%
·100%
Методическая ошибка, обусловлена
неколичественным выпадением осадка,
её устранить нельзя
Qоб= s-растворимость осадка
s-растворимость осадка
г/100мл воды, -объём
-объём
фильтрата, —
—
масса гравиметрической формы
Случайные ошибки
Относительное стандартное отклонение
 =
=
 -дисперсия
-дисперсия
массы гравиметрической формы
 -масса
-масса
гравиметрической формы
Ϭa1-погрешность
взвешивания тары
Ϭa2-погрешность
взвешивания тары с навеской
 =
= =0,0003
=0,0003
г Ϭa1= Ϭa2=0,0002г
Суммарная ошибка
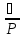 =
=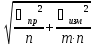
n-число проб
m-число измерений
 -погрешность
-погрешность
прибора
 -погрешность
-погрешность
измерения
8. Гравиметрическое
определение алюминия в каолине: этапы
определения, возможные формулы осадителей,
осаждаемой и гравиметрической формы,
механизм образования осадка, возможные
варианты загрязнения осадка, приемы
повышения чистоты осадка, погрешности
определения. Преимущества органических
осадителей. Условия аналитического
выделения осадков алюминия.
Механизм образования осадка:
В процессе образования осадка различают
три стадии :
1)образование зародышей кристаллов
2)рост кристаллов
3)объединение (агрегация) хаотично
ориентированных кристаллов
Насыщение=>пересыщение=>ПКИ>ПР=>
образование мельчайших зародышей
кристаллов
Осаждение происходит при определенной
степени пересыщения раствора
P= =
= s-растворимость,
s-растворимость, -относительное
-относительное
пересыщение,Q-концентрация
кристаллизующегося вещества в растворе
Центром кристалла может служить твердая
частица этого вещества или любая другая
твердая частица, которую мы вносим в
раствор, твердые частицы могут изначально
присутствовать в растворе как примесь.
Если осаждение происходи из разбавленных
растворов, то появление осадка занимает
время-индукцинный период.
В процессе добавления каждой новой
порции осадителя происходит мгновенное
пересыщение раствора, зародыши растут
быстро за счет окружающих их ионов, как
только зародыш дотиг определенного
размера выпадает осадок
Рост кристаллов идет параллельно 1-ой
стадии, происходит за счет диффузии
ионов к поверхности растущего кристалла.
Число и размер частиц осадка (дисперсность
системы кол-во в единицы объёма) зависит
от соотношения скоростей 1-ой и 2-ой
стадий
V1— скорость образования
зародышейV2-скорость
роста кристаллов
V1>>V2-мелкодисперсный
осадокV1<<V2-крупнокристаллический
осадок
Лимитирующую стадию определяет скорость
осаждения и концентрации ионов
При медленном осаждении лимитирующей
стадией является кристаллизация ,
частица окружена однородным слоем
осаждаемый ионов в результате получается
кристалл правильной форм
При высокой концентрации ионов
лимитирующей стадией становится диффузия
, образуются кристаллы не правильной
формы с большой площадью поверхности
Следует отметить, что на скорость
процесса кристаллизации влияет
 ,
,
влияние различно на скорость образования
различно на скорость образования
зародышей и на скорость роста кристаллов
В случае образования зародышей
V1=k·( экспоненциальный
экспоненциальный
закон
В случае роста кристаллов V2=k·
При высокой степени
 образуются
образуются
мелкодисперсные осадки, при уменьшении ,
,
образуются крупнокристаллические
осадки
Агрегация происходит в гетерогенной
системе, в значительной степени
определяется числом центров кристаллизации.
Чем больше центров кристаллизации , тем
в меньшей степени они укрупняются на
второй стадии , тем хуже структура и тем
выше дисперсность осадков.
К аналитическим свойствам осадка
относятся: растворимость, чистота,
фильтруемость.
Лучшими свойствами обладают
крупнокристаллические осадки.
Загрязнение осадков
В гарвиметрическом определении часто
возникают ошибки , вызванные переходом
осадка в раствор или веществ из раствора
в осадок-соосождение
Соосаждение происходит в процессе
образования осадка
Отрицательная роль : загрязнение осадка
Положительная роль :используется для
концентрирования микропримесей
Существует три типа соосаждения:
1)Адсорбция- соосаждение примесей на
поверхности уже сформированного осадка,
происходит в результате нескомпенсированности
зарядов внутри и на поверхности.
Характеризуется ярко выраженной
избирательность, преимущественно
адсорбируются те ионы, которые входят
в структуру осадка, противоионы-примеси
Адсорбция противоионов подчиняется
правилам Панета-Фаянса-Гана
А)при одинаковых концентрациях
адсорбируются многозарядные ионы
Б)при одинаковых зарядах адсорбируются
те, концентрация которых выше
В)при одинаковых концентрациях и
зарядах-те, которые образуют с ионами
решетки менее растворимое соединение
Г)в кислой среде соосаждение ионов
уменьшается в следствии конкурентной
адсорбции H3O+
Количество адсорбируемой примеси
зависит от величины поверхности осадка,
концентрации адсорбируемой примеси и
температуры ( с ↑ поверхности и ↑
концентрации- адсорбция ↑; с ↑ температуры
адсорбция ↓)
2)Окклюзия- загрязнение осадка в результате
захвата примеси внутрь растущего
кристалла, происходит в процессе
формирования осадка.
Различают 2-х видов: абсорбционная и
механическая
Механическа- случайный захват частиц
маточного раствора внутрь твердой фазы
вследствие нарушения механической
структуры
Характерна при выделении аморфных
осадков.
Окклюзированные примеси равномерно
распределены внутри, но не принимают
участие в построении решетки кристалла.
Адсорбционная-возникает при быстром
росте кристалла, когда ионы на поверхности
обратают кристаллизованным веществом.
Протекает вследствии адсорбции примесей
по микротрещинам кристаллической
структуры.
Окклюзия подчиняется тем же правилам,
что и адсорбция
Общие правила понижения окклюзии–замедление процесса выделения твердой
фазы-осаждение при малом пересыщении
, работают с разбавленными растворами
, осадитель добавляют по каплям, при
постоянном перемешивании.
3)изоморфное соосаждение характерно
для изоморфно кристаллизующегося
веществ, которые могут образовывать
смешанные кристаллы, примесь участвует
в построении кристаллической решетки,
наблюдается лишь в тех случаях, когда
вещества сходны по химическим свойствам
или ионы имеют одинаковые кч и радиус.
Совместное осаждение-выделение в твердую
фазу нескольких веществ, для которых в
услових осаждения достигнуты величины
их Kst
Последовательное осаждение- веделение
примеси на поверхности уже сформированного
осадка
Приемы и методы повышения чистоты
осадка
Зависят от типа соосаждения
1)адсорбционные примеси хорошо удаляются
промыванием осадка, более эффективно
многократное промывание малыми порциями
Выбор промывочной жидкости:
Не увеличивает растворимость осадка и
не ухудшает его фильтруемость, водой
промывают осадки с k~10-11/-12,
не подвергаемых пептизации, кристаллические
осадки с конст, растворимости 10-9/-11промывают разбавленным раствором
осадителя, аморфные осадки промывают
разбавленными растворами электролитов
коагуляторов, чтобы избежать пептизации
Промывние кристаллических осадков
проводят холодной промывочной жидкостью,
чтоб не увеличивать растворимость,
аморфные наоборот горячими
2)окклюзированные примеси , для избавления
от них:
Для кристаллических осадков-старение
Для аморфных-переосаждение
Погрешность гравиметрического
метода анализа
Общая погрешность анализа
Ϭ2=
 +
+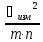
 -погрешность
-погрешность
пробоотбораm-число пробn-число параллельных
определений
 -погрешность
-погрешность
измерений
Результат находится по формуле
P,%= ·100%
·100%
Методическая ошибка, обусловлена
неколичественным выпадением осадка,
её устранить нельзя
Qоб= s-растворимость осадка
s-растворимость осадка
г/100мл воды, -объём
-объём
фильтрата, —
—
масса гравиметрической формы
Случайные ошибки
Относительное стандартное отклонение
 =
=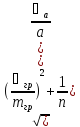
 -дисперсия
-дисперсия
массы гравиметрической формы
 -масса
-масса
гравиметрической формы
Ϭa1-погрешность
взвешивания тары
Ϭa2-погрешность
взвешивания тары с навеской
 =
= =0,0003
=0,0003
г Ϭa1= Ϭa2=0,0002г
Суммарная ошибка
 =
=
n-число проб
m-число измерений
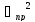 -погрешность
-погрешность
прибора
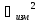 -погрешность
-погрешность
измерения
9. Гравиметрическое
определение железа в руде: этапы
определения, возможные формулы осадителя,
осаждаемой и гравиметрической формулы,
механизм образования коллоидной частицы,
процессы, приводящие к образованию
осадка, возможные варианты загрязнения
осадка, приемы повышения чистоты осадка,
погрешности. Условия аналитического
выделения осадков железа.
Гравиметрическое определение железа(III)
основано на его осаждении в виде
гидроксида железа(III)Fe(OH)3.
Трехвалентное железо осаждают раствором
аммиака, осаждаемой формой являетсяFe(OH)3.
Реакция:Fe(NO3)3+3NH3·H2O=Fe(OH)3+3NH4NO3.
При прокаливании гидроксид железа(III)
превращается в оксид железа(III),
который является гравиметрической
формой:Fe(OH)3=(t°)Fe2O3+3H2O.
Этапы определения:1) взятие навески
и ее растворение; 2) приготовление
раствора осадителя; 3) осаждение; 4)
фильтрование и промывание осадка; 5)
высушивание и прокаливание; 6) взвешивание
осадка, расчет содержания железа.
Расчет ведут по формулам


ωFe2O3=
 ,
,
ωFe
=

Механизм образования коллоидной
частицы:
Fe(NO3)3+3NH4OH(изб.)=Fe(OH)3↓+3NH4NO3
{[Fe(OH)3]m
· nOH—
·(n-x)NH4+}-x
·xNH4+
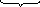


агрегат плотный слой
диффузный слой Мицелла
Ядро 
Коллоидная частица
Вещество в коллоидной системе имеет
большую развитую поверхность и
нескомпенсированный заряд на границе
разлела фаз. Существование
нескомпенсированного силового поля
ведет к адсорбции из раствора молекул
или ионов. Если коллоидная система
возникла в результате проведения
химической реакции осаждения, то частицы
адсорбируют в первую очередь те ионы,
которые могут достраивать кристаллическую
решетку. Адсорбированные ионы сообщают
частице «+» или «-« заряд. Слой
адсорбированных ионов на ядре – это
первичный адсорбционный слой. Заряд,
созданный таким слоем, достаточно высок
и обуславливает электростатическое
взаимодействие с иоами противоположного
знака. В результате образуется слой
противоионов, который выравнивает заряд
первичного слоя. Слой противоионов
имеет диффузный характер. Часть
противоионов, прочно связанных с
первичным слоем – это плотный слой,
остальные противоионы составляют
диффузный слой.
Образование осадкапроисходит
тогда, когда раствор становится
пересыщенным, т.е. [A+]m[B-]n>Ks(ПКИ>ПР). Образование осадков связано
с процессом укрупнения частиц, с
образованием кристаллической решетки
вещества. Этот процесс определяется
числом центров кристаллизации: чем
больше центров, тем в меньшей степени
они укрупняются и тем хуже структура и
выше дисперсность осадка.
Возможные варианты загрязнения:
1)Путем адсорбции ( для конкретного
примера хлорид-ионов на поверхности
осадка); 2)Окклюзия; 3)Изоморфное
соосаждение; 4) Совместное осаждение;
5) Последующее осаждение.
Приемы повышения чистоты осадка:
1) Адсорбированные на поверхности примеси
хорошо удаляются при промывании осадков
на фильтре при помощи промывных жидкостей,
т.к. примеси переходят в промывную
жидкость и уходят через поры фильтра.
Эффективно многократное промывание
небольшими порциями промывной жидкости.
Промывную жидкость выбирают максимально
тщательно, чтобы не увеличивать
растворимость осадка и не ухудшать его
фильтрацию. Кристаллические осадки
промывают холодными промывными
жидкостями, чтобы не увеличить
растворимость осадка, а аморфные –
наоборот горячими. Водой промывают
осадки с низкими константами растворимости
(ниже 10-11-10-12), а также те,
которые не подвергаются пептизации.
Если константа растворимости осадка
10-9-10-11и он кристаллический,
то его промывают разбавленным раствором
осадителя. Аморфные осадки промывают
разбавленными растворами
электролитов-коагулянтов (солиNH4+),
чтобы избежать пептизации(в опыте с
железом осадок промывали растворомNH4NO3).
Повышение температуры также способствует
уменьшению адсорбции (на конкретном
примере горячий раствор, содержащий
10% аммиак разбавляют горячей водой для
уменьшения адсорбции хлорид-ионов на
поверхности осадка). 2) Для очищения
окклюдированных примесей в случае
кристаллических осадков используют
старение, в случае аморфных осадков –
переосаждение.Степень окклюзии в
процессе осаждения можно уменьшить
медленным добавлением осадителя по
каплям, при перемешивании.
Погрешности:
1) Общая погрешность анализа σ2=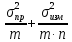 ,
,
где σпр2– погрешность
пробоотбора, σизм2–
погрешность измерения,m– число проб,n– число
параллельных определений.
2) Методическая ошибка OобOоб=
—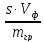 ,
,
гдеs– растворимость
осадка, г/100 мл воды;Vф
– объем фильтрата и промывных вод,
мл;mгр– масса
полученного осадка, г.
3) Относительное стандартное отклонение
 =
= , гдеσгр – дисперсия
, гдеσгр – дисперсия
массы гравиметрической формы;mгр– масса гравиметрической формы; σa– дисперсия массы исходной навески;a– масса исходной навески;p– процентное содержание вещества в
исследуемой пробе;n–число
измерений.
4) Погрешность взвешивания тары σa1и тары с навескойσa2σa1=σa2=0,0002
г, σгр= = 0,0003 г. 5) Относительное стандартное
= 0,0003 г. 5) Относительное стандартное
отклонение с учетом стадий пробоотбора
и пробоподготовки =
= , гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
, гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
Fe(OH)3– типичный пример осадка в аморфном
состоянии, легко дающий коллоидный
раствор.
Условия его осаждения следующие:
1)осаждение проводят из горячего раствора
анализируемого вещества горячим
раствором осадителя при перемешивании;
2)осаждение проводят из достаточно
концентрированного исследуемого
раствора концентрированным раствором
осадителя с последующим разбавлением(при
разбавлении устанавливается адсорбционное
равновесие, часть адсорбированных ионов
переходи в раствор, и осадок становится
более чистым); 3)осаждение проводят в
присутствии подходящего
электролита-коагулятора;
4)аморфные осадки почти не требуют
времени для созревания, их необходимо
фильтровать сразу после разбавления
раствора. Аморфные осадки нельзя
оставлять более, чем на несколько минут,
т.к. сильное уплотнение их затрудняет
последующее отмывание примесей, а также
при стоянии увеличивается количество
примесей, адсорбированных поверхностью
осадка.
10. Гравиметрическое определение никеля
в нихромовом сплаве: этапы определения,
возможные формулы осадителей, осаждаемой
и гравиметрической формулы, механизм
образования осадка, возможные варианты
загрязнения осадка, приемы повышения
чистоты осадка, погрешности. Условия
аналитического выделения осадков
никеля.
Гравиметрическое определение никеля
в нихромовом сплаве основано на его
осаждении в виде диметилглиоксимата
никеля Ni(HDMG)2.
Никель осаждают 1 %-ным спиртовым раствором
диметикглиоксимаH2DMG,
осаждаемой формой являетсяNi(HDMG)2.
Реакция:Ni2++2H2DMG=Ni(HDMG)2+2H+.
После высушивания осадка остается сухойNi(HDMG)2,
который является гравиметрической
формой.
Этапы определения:1) взятие навески
и ее растворение; 2) приготовление
раствора осадителя; 3) осаждение; 4)
фильтрование и промывание осадка; 5)
высушивание; 6) взвешивание осадка,
расчет содержания никеля.
Расчет ведут по формуле ωNi=

Механизм образования осадка:в
процессе образования осадка различают
3 параллельных процесса: 1) образование
зародышей кристалла (центров
кристаллизации); 2) рост кристаллов; 3)
объединение (агрегация) хаотично
ориентированных мелких кристаллов. В
начальный момент происходит насыщение
раствора, а затем его пересыщение. В
момент определенной пересыщенности
раствора, начинается выпадение
осадка.Центром кристалла может служить
твердая частица этого вещества или
любая другая твердая частица, которую
мы вносим в раствор, твердые частицы
могут изначально присутствовать в
растворе как примесь.
Если осаждение происходит из разбавленных
растворов, то появление осадка занимает
время-индукционный период.
В процессе добавления каждой новой
порции осадителя происходит мгновенное
пересыщение раствора, зародыши растут
быстро за счет окружающих их ионов, как
только зародыш достиг определенного
размера выпадает осадок.
Рост кристаллов идет параллельно 1-ой
стадии, происходит за счет диффузии
ионов к поверхности растущего кристалла.
Число и размер частиц осадка (дисперсность
системы кол-во в единицы объёма) зависит
от соотношения скоростей 1-ой и 2-ой
стадий (V1— скорость
образования зародышей,V2-скорость
роста кристаллов):V1>>V2-мелкодисперсный
осадок,V1<<V2-крупнокристаллический
осадок. Какая из стадий будет лимитировать
определяет скорость осаждения и
концентрации ионов. При медленном
осаждении лимитирующей стадией является
кристаллизация, частица окружена
однородным слоем осаждаемых ионов в
результате получается кристалл правильной
формы. При высокой концентрации ионов
лимитирующей стадией становится
диффузия, образуются кристаллы
неправильной формы с большой площадью
поверхности. Следует отметить, что на
скорость процесса кристаллизации влияет ,
,
влияние различно на скорость образования
различно на скорость образования
зародышей и на скорость роста кристаллов.
При высокой степени образуются
образуются
мелкодисперсные осадки, при уменьшении образуются крупнокристаллические
образуются крупнокристаллические
осадки. Агрегация происходит в гетерогенной
системе, в значительной степени
определяется числом центров
кристаллизации.Чем больше центров
кристаллизации, тем в меньшей степени
они укрупняются на второй стадии, тем
хуже структура и тем выше дисперсность
осадков.
К аналитическим свойствам осадка
относятся: растворимость, чистота,
фильтруемость.Лучшими свойствами
обладают крупнокристаллические осадки.
Возможные варианты загрязнения: 1)
Путем адсорбции ( для конкретного примера
хлорид-ионов на поверхности осадка); 2)
Окклюзия; 3) Изоморфное соосаждение; 4)
Совместное осаждение; 5) Последующее
осаждение.
Приемы повышения чистоты осадка:
1) Адсорбированные на поверхности примеси
хорошо удаляются при промывании осадков
на фильтре при помощи промывных жидкостей,
т.к. примеси переходят в промывную
жидкость и уходят через поры фильтра.
Эффективно многократное промывание
небольшими порциями промывной жидкости.
Промывную жидкость выбирают максимально
тщательно, чтобы не увеличивать
растворимость осадка и не ухудшать его
фильтрацию. Кристаллические осадки
промывают холодными промывными
жидкостями, чтобы не увеличить
растворимость осадка, а аморфные –
наоборот горячими. Водой промывают
осадки с низкими константами растворимости
(ниже 10-11-10-12), а также те,
которые не подвергаются пептизации.
Если константа растворимости осадка
10-9-10-11и он кристаллический,
то его промывают разбавленным раствором
осадителя. Аморфные осадки промывают
разбавленными растворами
электролитов-коагулянтов (солиNH4+),
чтобы избежать пептизации (в опыте с
железом осадок промывали растворомNH4NO3).
Повышение температуры также способствует
уменьшению адсорбции (на конкретном
примере горячий раствор, содержащий
10% аммиак разбавляют горячей водой для
уменьшения адсорбции хлорид-ионов на
поверхности осадка). 2) Для очищения
окклюдированных примесей в случае
кристаллических осадков используют
старение, в случае аморфных осадков –
переосаждение.Степень окклюзии в
процессе осаждения можно уменьшить
медленным добавлением осадителя по
каплям, при перемешивании.
Погрешности:1) Общая погрешность
анализа σ2=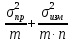 ,
,
где σпр2– погрешность
пробоотбора, σизм2–
погрешность измерения,m– число проб,n– число
параллельных определений.
2) Методическая ошибка OобOоб=
— ,
,
гдеs– растворимость
осадка, г/100 мл воды;Vф
– объем фильтрата и промывных вод,
мл;mгр– масса
полученного осадка, г.
3) Относительное стандартное отклонение
 =
= , гдеσгр – дисперсия
, гдеσгр – дисперсия
массы гравиметрической формы;mгр– масса гравиметрической формы; σa– дисперсия массы исходной навески;a– масса исходной навески;p– процентное содержание вещества в
исследуемой пробе;n–число
измерений.
4) Погрешность взвешивания тары σa1и тары с навескойσa2σa1=σa2=0,0002
г, σгр= = 0,0003 г.
= 0,0003 г.
5) Относительное стандартное отклонение
с учетом стадий пробоотбора и
пробоподготовки
 =
= , гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
, гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
Ni(HDMG)2– кристаллический осадок.
Условия его осаждения следующие:
1) осаждение ведут из достаточно
разбавленного исследуемого раствора
разбавленным раствором осадителя
(концентрации исследуемого раствора и
раствора осадителя должны быть примерно
одинаковыми);
2) раствор осадителя прибавляют медленно,
по каплям, при постоянном перемешивании
стеклянной палочкой (это предотвращает
явление окклюзии);
3) осаждение ведут из подогретого
исследуемого раствора горячим раствором
осадителя (для предотвращения пептизации);
4) к раствору прибавляют вещества,
способствующие повышению растворимости
осадка (увеличивают Iраствора), а затем понижают его
растворимость путем прибавления избытка
осадителя;
5) осадок оставляют на «созревание».
11. Гравиметрическое определение меди:
этапы определения, возможные формулы
осадителей, осаждаемой и гравиметрической
формулы, механизм образования осадка,
возможные варианты загрязнения осадка,
приемы повышения чистоты осадка,
погрешности. Преимущества органических
осадителей. Условия выделения осадков.
При гравиметрическом определении меди
медь из раствора осаждают различными
осадителями: 1) раствор аммиака осаждает
из нагретого раствора осадок Cu(OH)2;
2) Тиокарбонат калияK2CS3осаждает из нагретого раствора осадокCuS, который сушат при
100-110 ;
;
3) В виде оксалата медь осаждается в
присутствиеCH3COOH;
4) При определении меди в виде
тетророданомеркуриатамедиCu[Hg(SCN)4]
медь осаждают из нагретого до кипения
раствора содержащего серную или азотную
кислоту, действиемK2[Hg(SCN)4].
Метод рекомендован для определения
меди в медных рудах; 5) Соль Рейнеке
(тетрароданодиаминохромат аммония)
NH4[Cr(NH3)2(SCN)4]
является избирательным реагентом для
определения меди в присутствие многих
посторонних ионов. Осаждение проводят
как в кислом, так и в аммиачном растворе
в виде [Cu(NH3)4][Cr(NH3)2(SCN)4]2
после предварительного восстановления
меди до одновалентного состояния
оловом(II). Для осаждения меди используются
также различные органические реагенты:
1) 8- оксихинолин осаждает медь в
уксуснокислом, аммиачном и щелочном
растворах при pH=5.33 — 14.55. Осадок, высушенный
при 105-110°С, соответствует составу
Cu(C9H6ON)2; 2) Медь осаждается
спиртовым раствором β-бензоиноксима в
слабощелочной среде в виде хлопьевидного
зеленого осадка составаCu(C6H5CHOCNOC6H5)2.
Осадок высушивают при 105-110 ;
;
3) Салицилальдиоксим осаждает Cu (II) в
виде внутрикомплексного соединения
Cu(C7H6O2N)2в
уксуснокислой среде, среде ацетатного
буфера или ацетата аммония; 4) При действии
купферона наCu(II)
образуется купферонат меди (II)
с формулой Cu(C6H5N(NO)O)2;
5) При действии глицина на медь образуется
кристаллический осадок глицината меди
(II)Cu(NH2CH2COO)2.
Рассмотрим гравиметрическое определение
меди на примере осаждения ее
глицином.Реакция:CuO+2NH2CH2COOH=Cu(NH2CH2COO)2+H2OВданном случае глицинNH2CH2COOHявляется
осадителем, глицинат меди (II)Cu(NH2CH2COO)2– осаждаемой формой. При высушивании
получается гравиметрическая форма
сухогоCu(NH2CH2COO)2.
Этапы определения:1) взятие навески
и её растворение;2) приготовление раствора
осадителя;3) осаждение;4) фильтрование
и промывание;5) высушивание осадка;6)
взвешивание осадка, расчёт содержания
меди.
Механизм образования осадка:в
процессе образования осадка различают
3 параллельных процесса: 1) образование
зародышей кристалла (центров
кристаллизации); 2) рост кристаллов; 3)
объединение (агрегация) хаотично
ориентированных мелких кристаллов. В
начальный момент происходит насыщение
раствора, а затем его пересыщение. В
момент определенной пересыщенности
раствора, начинается выпадение осадка.
Центром кристалла может служить твердая
частица этого вещества или любая другая
твердая частица, которую мы вносим в
раствор, твердые частицы могут изначально
присутствовать в растворе как примесь.
Если осаждение происходит из разбавленных
растворов, то появление осадка занимает
время-индукционный период.
В процессе добавления каждой новой
порции осадителя происходит мгновенное
пересыщение раствора, зародыши растут
быстро за счет окружающих их ионов, как
только зародыш достиг определенного
размера выпадает осадок.
Рост кристаллов идет параллельно 1-ой
стадии, происходит за счет диффузии
ионов к поверхности растущего кристалла.
Число и размер частиц осадка (дисперсность
системы кол-во в единицы объёма) зависит
от соотношения скоростей 1-ой и 2-ой
стадий (V1— скорость
образования зародышей,V2-скорость
роста кристаллов):V1>>V2-мелкодисперсный
осадок,V1<<V2-крупнокристаллический
осадок. Какая из стадий будет лимитировать
определяет скорость осаждения и
концентрации ионов. При медленном
осаждении лимитирующей стадией является
кристаллизация, частица окружена
однородным слоем осаждаемых ионов в
результате получается кристалл правильной
формы. При высокой концентрации ионов
лимитирующей стадией становится
диффузия, образуются кристаллы
неправильной формы с большой площадью
поверхности. Следует отметить, что на
скорость процесса кристаллизации влияет ,
,
влияние различно на скорость образования
различно на скорость образования
зародышей и на скорость роста кристаллов.
При высокой степени образуются
образуются
мелкодисперсные осадки, при уменьшении образуются крупнокристаллические
образуются крупнокристаллические
осадки. Агрегация происходит в гетерогенной
системе, в значительной степени
определяется числом центров
кристаллизации.Чем больше центров
кристаллизации, тем в меньшей степени
они укрупняются на второй стадии, тем
хуже структура и тем выше дисперсность
осадков.
К аналитическим свойствам осадка
относятся: растворимость, чистота,
фильтруемость.Лучшими свойствами
обладают крупнокристаллические осадки.
Возможные варианты загрязнения: 1)
Путем адсорбции ( для конкретного примера
хлорид-ионов на поверхности осадка); 2)
Окклюзия; 3) Изоморфное соосаждение; 4)
Совместное осаждение; 5) Последующее
осаждение.
Приемы повышения чистоты осадка:
1) Адсорбированные на поверхности примеси
хорошо удаляются при промывании осадков
на фильтре при помощи промывных жидкостей,
т.к. примеси переходят в промывную
жидкость и уходят через поры фильтра.
Эффективно многократное промывание
небольшими порциями промывной жидкости.
Промывную жидкость выбирают максимально
тщательно, чтобы не увеличивать
растворимость осадка и не ухудшать его
фильтрацию. Кристаллические осадки
промывают холодными промывными
жидкостями, чтобы не увеличить
растворимость осадка, а аморфные –
наоборот горячими. Водой промывают
осадки с низкими константами растворимости
(ниже 10-11-10-12), а также те,
которые не подвергаются пептизации.
Если константа растворимости осадка
10-9-10-11и он кристаллический,
то его промывают разбавленным раствором
осадителя. Аморфные осадки промывают
разбавленными растворами
электролитов-коагулянтов (солиNH4+),
чтобы избежать пептизации (в опыте с
железом осадок промывали растворомNH4NO3).
Повышение температуры также способствует
уменьшению адсорбции (на конкретном
примере горячий раствор, содержащий
10% аммиак разбавляют горячей водой для
уменьшения адсорбции хлорид-ионов на
поверхности осадка). 2) Для очищения
окклюдированных примесей в случае
кристаллических осадков используют
старение, в случае аморфных осадков –
переосаждение.Степень окклюзии в
процессе осаждения можно уменьшить
медленным добавлением осадителя по
каплям, при перемешивании.
Погрешности:1) Общая погрешность
анализа σ2 =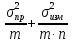 ,
,
где σпр2– погрешность
пробоотбора, σизм2–
погрешность измерения,m– число проб,n– число
параллельных определений
2) Методическая ошибка OобOоб=
—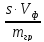 ,
,
гдеs– растворимость
осадка, г/100 мл воды;Vф
– объем фильтрата и промывных вод,
мл;mгр– масса
полученного осадка, г.
3) Относительное стандартное отклонение
 =
= , гдеσгр – дисперсия
, гдеσгр – дисперсия
массы гравиметрической формы;mгр– масса гравиметрической формы; σa– дисперсия массы исходной навески;a– масса исходной навески;p– процентное содержание вещества в
исследуемой пробе;n–число
измерений.
4) Погрешность взвешивания тары σa1и тары с навескойσa2σa1=σa2=0,0002
г, σгр= = 0,0003 г.
= 0,0003 г.
5) Относительное стандартное отклонение
с учетом стадий пробоотбора и
пробоподготовки
 =
= , гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
, гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
Преимущества органических осадителей:
1. Пользуясь органическими осадителями,
можно осаждать и разделять различные
элементы из очень сложных смесей.
Например, при помощи диметилглиоксима
возможно количественное осаждение
катионов никеля в присутствии многих
других катионов.
2. Осадки, получающиеся с органическими
осадителями, хорошо отфильтровываются
и промываются (например, осадки комплексных
соединений катионов, содержащих в
качестве лигандов пиридин или другие
органические соединения). Это дает
возможность легко отмывать от осадков
примеси, содержащиеся в анализируемом
растворе.
3. Осадки, получающиеся при действии на
катионы или анионы органических
осадителей, отличаются большим
молекулярным весом. Вследствие этого
точность анализа повышается. Например,
определение магния, алюминия и других
катионов проводится с большой точностью
осаждением их в виде оксихинолятов,
обладающих большим молекулярным весом.
4. В составе осадков, являющихся
соединениями неорганических веществ
с органическими компонентами, обычно
содержится мало соосаждающихся пиримесей.
Cu(NH2CH2COO)2– кристаллический осадок, поэтому
условия его выделения следующие:
1) осаждение ведут из достаточно
разбавленного исследуемого раствора
разбавленным раствором осадителя
(концентрации исследуемого раствора и
раствора осадителя должны быть примерно
одинаковыми);
2) раствор осадителя прибавляют медленно,
по каплям, при постоянном перемешивании
стеклянной палочкой (это предотвращает
явление окклюзии);
3) осаждение ведут из подогретого
исследуемого раствора горячим раствором
осадителя (для предотвращения пептизации);
4) к раствору прибавляют вещества,
способствующие повышению растворимости
осадка (увеличивают Iраствора), а затем понижают его
растворимость путем прибавления избытка
осадителя;
5) осадок оставляют на «созревание».
12. Гравиметрическое определение
кремния в силикатных породах: этапы
определения, возможные формулы осадителя,
осаждаемой и гравиметрической формулы,
механизм образования коллоидной частицы,
процессы, приводящие к образованию
осадка, возможные варианты загрязнения
осадка, приемы повышения чистоты осадка,
погрешности. Классификация коллоидных
систем. Условия аналитического выделения
кремнекислоты.
При гравиметрическом определении
кремния растворимый силикат натрия
Na2SiO3,
полученный в результате сплавления не
разлагаемой кремниевой кислоты с содойNa2CO3,
обрабатывается сильной кислотойHCl.
Реакция:Na2SiO3+2HCl=H2SiO3↓+2NaCl.
Осадителем в данном случае являетсяHCl, осаждаемой формой –H2SiO3.
При высушивании и прокаливании получается
гравиметрическая формаSiO2.
Этапы определения:1) взятие навески
и ее растворение; 2) приготовление
раствора осадителя; 3) осаждение; 4)
фильтрование и промывание осадка; 5)
высушивание и прокаливание осадка;; 6)
взвешивание осадка, расчет содержания
кремния.
Механизм образования коллоидной
частицы: Вещество в коллоидной системе
имеет большую развитую поверхность и
нескомпенсированный заряд на границе
разлела фаз. Существование
нескомпенсированного силового поля
ведет к адсорбции из раствора молекул
или ионов. Если коллоидная система
возникла в результате проведения
химической реакции осаждения, то частицы
адсорбируют в первую очередь те ионы,
которые могут достраивать кристаллическую
решетку. Адсорбированные ионы сообщают
частице “+» или “-“ заряд. Слой
адсорбированных ионов на ядре – это
первичный адсорбционный слой. Заряд,
созданный таким слоем, достаточно высок
и обуславливает электростатическое
взаимодействие с иоами противоположного
знака. В результате образуется слой
противоионов, который выравнивает заряд
первичного слоя. Слой противоионов
имеет диффузный характер. Часть
противоионов, прочно связанных с
первичным слоем – это плотный слой,
остальные противоионы составляют
диффузный слой.
Образование осадкапроисходит
тогда, когда раствор становится
пересыщенным, т.е. [A+]m[B-]n>Ks(ПКИ>ПР). Образование осадков связано
с процессом укрупнения частиц, с
образованием кристаллической решетки
вещества. Этот процесс определяется
числом центров кристаллизации: чем
больше центров, тем в меньшей степени
они укрупняются и тем хуже структура и
выше дисперсность осадка.
Возможные варианты загрязнения:1)
Путем адсорбции ( для конкретного примера
хлорид-ионов на поверхности осадка); 2)
Окклюзия; 3) Изоморфное соосаждение; 4)
Совместное осаждение; 5) Последующее
осаждение.
Приемы повышения чистоты осадка:
1) Адсорбированные на поверхности примеси
хорошо удаляются при промывании осадков
на фильтре при помощи промывных жидкостей,
т.к. примеси переходят в промывную
жидкость и уходят через поры фильтра.
Эффективно многократное промывание
небольшими порциями промывной жидкости.
Промывную жидкость выбирают максимально
тщательно, чтобы не увеличивать
растворимость осадка и не ухудшать его
фильтрацию. Кристаллические осадки
промывают холодными промывными
жидкостями, чтобы не увеличить
растворимость осадка, а аморфные –
наоборот горячими. Водой промывают
осадки с низкими константами растворимости
(ниже 10-11-10-12), а также те,
которые не подвергаются пептизации.
Если константа растворимости осадка
10-9-10-11и он кристаллический,
то его промывают разбавленным раствором
осадителя. Аморфные осадки промывают
разбавленными растворами
электролитов-коагулянтов (солиNH4+),
чтобы избежать пептизации (в опыте с
железом осадок промывали растворомNH4NO3).
Повышение температуры также способствует
уменьшению адсорбции (на конкретном
примере горячий раствор, содержащий
10% аммиак разбавляют горячей водой для
уменьшения адсорбции хлорид-ионов на
поверхности осадка). 2) Для очищения
окклюдированных примесей в случае
кристаллических осадков используют
старение, в случае аморфных осадков –
переосаждение.Степень окклюзии в
процессе осаждения можно уменьшить
медленным добавлением осадителя по
каплям, при перемешивании.
Погрешности:
1) Общая погрешность анализа σ2 = ,
,
где σпр2– погрешность
пробоотбора, σизм2–
погрешность измерения,m– число проб,n– число
параллельных определений.
2) Методическая ошибка OобOоб=
— ,
,
гдеs– растворимость
осадка, г/100 мл воды;Vф
– объем фильтрата и промывных вод,
мл;mгр– масса
полученного осадка, г.
3) Относительное стандартное отклонение
 =
= , гдеσгр – дисперсия
, гдеσгр – дисперсия
массы гравиметрической формы;mгр– масса гравиметрической формы; σa– дисперсия массы исходной навески;a– масса исходной навески;p– процентное содержание вещества в
исследуемой пробе;n–число
измерений.
4) Погрешность взвешивания тары σa1и тары с навескойσa2σa1=σa2=0,0002
г, σгр= = 0,0003 г.
= 0,0003 г.
5) Относительное стандартное отклонение
с учетом стадий пробоотбора и
пробоподготовки
 =
= , гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
, гдеn– число проб;m– число параллельных измерений; σпр2– погрешность пробоотбора; σизм2– погрешность измерения.
Классификация коллоидных систем. В
зависимости от характера межмолекулярных
сил, которые действуют на границе раздела
фаз коллоидные растворы делят на
лиофильные и лиофобные. Вокруг лиофильной
частицы располагается прочная сольватная
оболочка. В этих оболочках молекулы
ориентированы определенным образом и
образуют более или менее правильные
структуры. Вокруг лиофобной частицы
раствора также имеются сольватные
оболочки, но они непрочные и не предохраняют
молекулы от слипания.
H2SiO3– аморфный осадок, поэтому
условия его осаждения следующие:
1)осаждение проводят из горячего раствора
анализируемого вещества горячим
раствором осадителя при перемешивании;
2)осаждение проводят из достаточно
концентрированного исследуемого
раствора концентрированным раствором
осадителя с последующим разбавлением(при
разбавлении устанавливается адсорбционное
равновесие, часть адсорбированных ионов
переходи в раствор, и осадок становится
более чистым); 3)осаждение проводят в
присутствии подходящего
электролита-коагулятора;
4)аморфные осадки почти не требуют
времени для созревания, их необходимо
фильтровать сразу после разбавления
раствора. Аморфные осадки нельзя
оставлять более, чем на несколько минут,
т.к. сильное уплотнение их затрудняет
последующее отмывание примесей, а также
при стоянии увеличивается количество
примесей, адсорбированных поверхностью
осадка.
Автор:
Sara Rhodes
Дата создания:
11 Февраль 2021
Дата обновления:
21 Сентябрь 2023

Содержание
- Ключевые выводы
- Пример случайной ошибки и причины
- Пример систематической ошибки и причины
- Ключевые выводы: случайная ошибка против систематической ошибки
- Источники

Независимо от того, насколько вы осторожны, в измерениях всегда есть погрешности. Ошибка — это не «ошибка», это часть процесса измерения. В науке ошибка измерения называется ошибкой эксперимента или ошибкой наблюдения.
Есть два широких класса ошибок наблюдения: случайная ошибка а также систематическая ошибка. Случайная погрешность непредсказуемо меняется от одного измерения к другому, в то время как систематическая ошибка имеет одинаковое значение или пропорцию для каждого измерения. Случайные ошибки неизбежны, но они группируются вокруг истинного значения. Систематической ошибки часто можно избежать путем калибровки оборудования, но если ее не исправить, результаты измерений могут отличаться от истинного значения.
Ключевые выводы
- Случайная погрешность приводит к тому, что одно измерение немного отличается от следующего. Это происходит из-за непредсказуемых изменений во время эксперимента.
- Систематическая ошибка всегда влияет на измерения в одинаковой степени или в одинаковой пропорции, при условии, что показания снимаются каждый раз одинаково. Это предсказуемо.
- Случайные ошибки нельзя исключить из эксперимента, но можно уменьшить большинство систематических ошибок.
Пример случайной ошибки и причины
Если вы выполните несколько измерений, значения сгруппируются вокруг истинного значения. Таким образом, случайная ошибка в первую очередь влияет на точность. Обычно случайная ошибка влияет на последнюю значащую цифру измерения.
Основными причинами случайной ошибки являются ограниченность инструментов, факторы окружающей среды и небольшие отклонения в процедуре. Например:
- Взвешивая себя на весах, вы каждый раз позиционируете себя немного по-другому.
- При измерении объема в колбе вы можете каждый раз считывать значение под другим углом.
- Измерение массы образца на аналитических весах может дать разные значения, поскольку потоки воздуха влияют на весы или когда вода входит и выходит из образца.
- На измерение вашего роста влияют незначительные изменения позы.
- Измерение скорости ветра зависит от высоты и времени проведения измерения. Необходимо снять несколько показаний и усреднить, потому что порывы ветра и изменения направления влияют на значение.
- Показания должны оцениваться, когда они попадают между отметками на шкале или когда принимается во внимание толщина измерительной отметки.
Поскольку случайная ошибка возникает всегда и не может быть предсказана, важно взять несколько точек данных и усреднить их, чтобы получить представление о величине вариации и оценить истинное значение.
Пример систематической ошибки и причины
Систематическая ошибка предсказуема и либо постоянна, либо пропорциональна измерению. Систематические ошибки в первую очередь влияют на точность измерения.
Типичные причины систематической ошибки включают ошибку наблюдений, несовершенную калибровку прибора и влияние окружающей среды. Например:
- Если вы забываете тарировать или обнулять весы, измерения массы всегда отклоняются на одну и ту же величину. Ошибка, вызванная тем, что прибор не установил ноль перед использованием, называется ошибкой. ошибка смещения.
- Отсутствие показаний мениска на уровне глаз для измерения объема всегда приведет к неточным показаниям. Значение будет постоянно низким или высоким, в зависимости от того, отсчитывается ли отсчет от отметки выше или ниже.
- Измерение длины металлической линейкой даст другой результат при низкой температуре, чем при высокой температуре, из-за теплового расширения материала.
- Неправильно откалиброванный термометр может давать точные показания в определенном температурном диапазоне, но становиться неточными при более высоких или более низких температурах.
- Расстояние между новой тканевой рулеткой и старой натянутой лентой отличается. Пропорциональные погрешности этого типа называются ошибки масштабного коэффициента.
- Дрейф происходит, когда последовательные показания со временем становятся стабильно ниже или выше. Электронное оборудование склонно к дрейфу. Многие другие инструменты подвержены дрейфу (обычно положительному) по мере того, как устройство нагревается.
После выявления причины систематическая ошибка может быть в некоторой степени уменьшена. Систематические ошибки можно свести к минимуму, регулярно калибруя оборудование, используя контроли в экспериментах, прогревая инструменты перед снятием показаний и сравнивая значения со стандартами.
Хотя случайные ошибки можно минимизировать за счет увеличения размера выборки и усреднения данных, систематическую ошибку компенсировать сложнее. Лучший способ избежать систематической ошибки — это знать ограничения инструментов и иметь опыт их правильного использования.
Ключевые выводы: случайная ошибка против систематической ошибки
- Двумя основными типами ошибок измерения являются случайная ошибка и систематическая ошибка.
- Случайная погрешность приводит к тому, что одно измерение немного отличается от следующего. Это происходит из-за непредсказуемых изменений во время эксперимента.
- Систематическая ошибка всегда влияет на измерения в одинаковой степени или в одинаковой пропорции, при условии, что показания снимаются каждый раз одинаково. Это предсказуемо.
- Случайные ошибки нельзя исключить из эксперимента, но можно уменьшить большинство систематических ошибок.
Источники
- Блэнд, Дж. Мартин и Дуглас Г. Альтман (1996). «Статистические заметки: ошибка измерения». BMJ 313.7059: 744.
- Кокран, У. Г. (1968). «Ошибки измерения в статистике». Технометрика. Taylor & Francis, Ltd. от имени Американской статистической ассоциации и Американского общества качества. 10: 637–666. DOI: 10.2307 / 1267450
- Додж, Ю. (2003). Оксфордский словарь статистических терминов. ОУП. ISBN 0-19-920613-9.
- Тейлор, Дж. Р. (1999). Введение в анализ ошибок: исследование неопределенностей в физических измерениях. Книги университетских наук. п. 94. ISBN 0-935702-75-X.