Содержание
«Такие множественные пороки — это, скорее всего, генетика»
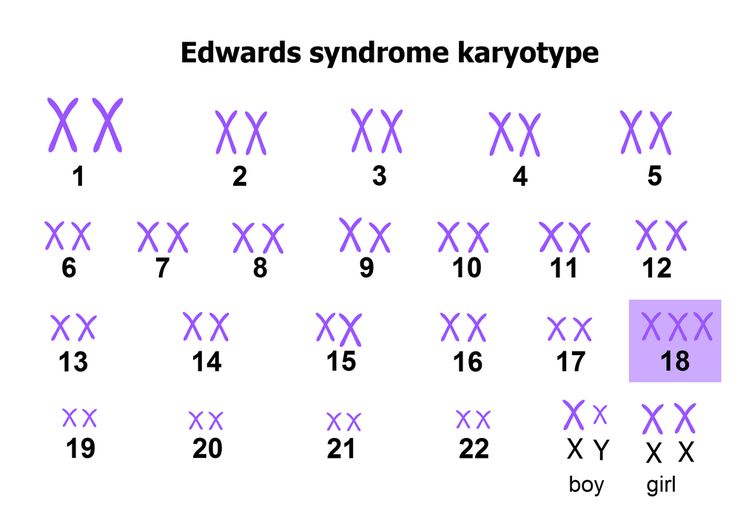
Что такое синдром Эдвардса
Как диагностируют синдром Эдвардса
«Моя дочь растет, развивается, она личность и просто ребенок»
Программа перинатальной паллиативной помощи
Светлая печаль, как сама жизнь
У меня еще есть время, чтобы быть рядом
Информация про синдром Эдвардса на всех медицинских ресурсах неутешительная: прогноз для этого заболевания весьма неблагоприятный. Но вопреки заложенному сценарию развития заболевания многие дети при хорошем уходе и паллиативной помощи живут довольно долго. Лекарство от заболевания одно — родительская любовь.
«Такие множественные пороки — это, скорее всего, генетика»
Саше Глаголевой поставили синдром Эдвардса вскоре после рождения. Сейчас Саше 8 лет, хотя во всех медицинских справочниках написано, что девочки с таким заболеванием живут в среднем около 10 месяцев, а мальчики от двух до трех месяцев, и вообще около 90% детей с синдромом Эдвардса умирают в течение первого года жизни.
Саша родилась со множественными пороками развития, и врачи в реанимации сразу предположили генетическое заболевание.
Родители не могли поверить, ведь и сами они были здоровы, и у них уже был совершенно здоровый ребенок, но врачи объяснили, что дети с генетическими проблемами иногда появляются из-за спонтанной мутация в генах или хромосомах, а не только из-за наследственности.
На третий день генетический тест подтвердил предположение врачей — синдром Эдвардса.
Что такое синдром Эдвардса
Синдром Эдвардса, или трисомияXтрисомияТрисомия означает вариант хромосомной мутации, при которой в клетках человека содержится не 46, а 47 хромосом. по 18-ой хромосоме — это генетическое заболевание, при котором ребенок наследует три восемнадцатых хромосомы вместо двух. Мутация происходит в результате нерасхождения 18-ой хромосомы во время деления клеток.
В описаниях заболевания сказано, что в 60% случаях из-за пороков, несовместимых с жизнью, дети с синдромом Эдвардса погибают внутриутробно. В группе трисомий только в трех случаях — при синдроме Эдвардса, синдроме Дауна (трисомия 21-ой хромосомы) и синдроме Патау (трисомия 13-ой хромосомы) возможно рождение живого ребенка и его дальнейшее, хоть и осложненное, развитие. При других вариантах добавочных хромосом патология несовместима с жизнью.
Распространенность синдрома Эдвардса в мире составляет один случай на 3 000-7 000 новорожденных, определенной зависимости от региона или расы нет. Чаще синдром встречается у девочек. Ребенок с трисомией 18-ой хромосомы может родиться у любой женщины, но считается, что риск усиливается с возрастом матери. А некоторые исследования показали, что в последние годы синдром Эдвардса имеет тенденцию к увеличению. Возможно, это связано с улучшением диагностики и с повышением возраста рожающих женщин.
Хотя основные симптомы заболевания описаны еще в начале XX века, первый полный обзор синдрома и его основной причины — появление лишней 18-ой хромосомы — был сделан только в 1960 году британским медицинским генетиком Джоном Эдвардсом, в честь которого и назвали эту патологию.
Заболевание сопровождается многочисленными нарушениями в развитии систем органов, но если лишняя 18-ая хромосома находится не в каждой клетке организма, что встречается у 5% людей с этой патологией и называется мозаицизмом, синдром Эдвардса может быть более легкой формы.
Как и другие заболевания с хромосомными мутациями, синдром Эдвардса неизлечим. В настоящее время лечение состоит, в основном, из паллиативной помощи.
Как диагностируют синдром Эдвардса
Заподозрить наличие синдрома Эдвардса можно на УЗИ плода и допплерографии по косвенным признакам: недоразвитию одной из пупочных артерий, многоводию, уменьшению размеров плаценты. Но ультразвуковое исследование может показать только характерные признаки патологии, оно не может подтвердить, что есть именно этот синдром.

Эпоха АлександрыЕвгений Глаголев, отец тяжелобольной девочки рассказывает о том, как изменилась жизнь его семьи, почему он решил сменить профессию и зачем родителям время от времени выезжать куда-то вдвоем
Вообще, для диагностики заболевания большое значение имеет совокупность факторов: результаты лабораторного скрининга, данные УЗИ, срок беременности и возраст матери. Если есть сразу несколько симптомов необходимо провести дополнительные инвазивные исследования — биопсию ворсин хориона (анализ клеток эмбриональной ткани), амниоцентез (взятие на анализ околоплодных вод) или кордоцентез (взятие анализа крови из пуповины). Полученный в ходе процедур материал плода отправляется на кариотипирование — анализ на выявление нарушений хромосомного набора.
У новорожденного ребенка для определения точной причины врожденных дефектов тоже должно быть проведено хромосомное исследование с помощью анализа крови.
«Моя дочь растет, развивается, она личность и просто ребенок»
Екатерина С., мама восьмилетней дочери с полной формой трисомии по 18-ой хромосоме считает, что и медики, и генетики очень мало знают о жизни и развитии детей с этим диагнозом.
После того, как ее дочери поставили синдром Эдвардса, Екатерина оказалась буквально в информационном вакууме, и все, что она прочитала о болезни, было очень пессимистичным относительно прогноза жизни таких детей. Она начала общаться с другими родителями детей с трисомией и решила объединить их в сообщество, в котором сейчас более 40 семей.
По ее мнению, данные в справочниках по генетике по этому заболеванию устарели — они написаны 20-30 лет назад, а ведь за это время медицина шагнула далеко вперед, и выхаживание детей с синдромом Эдвардса стало возможным. Она понимает, что вылечить синдром нереально, но уверена, что облегчить состояние ребенка можно.
Екатерина рассказывает, что в регистре их сообщества 79% детей с полной трисомией и 21% с мозаичной формой, их возраст — от года до 10 лет. Также она сообщила, что по данным исследований группы ученых во главе с американским профессором Робертом Мейером (Robert E Meyer), около 12,3% детей с трисомией по 18-ой хромосоме доживают до пяти лет.

Каждая секунда имеет значение и смыслИстория Ани и Славы Черепановых. Родителей, потерявших двоих детей
По мнению Екатерины, это свидетельствует о том, что несмотря на тяжесть состояния и высокую смертность, синдром Эдвардса некорректно рассматривать как летальное и несовместимое с жизнью заболевание: «Моя дочь растет, развивается, она личность и просто ребенок, хоть и не нормотипичный, но со своим характером и потребностями».
Программа перинатальной паллиативной помощи
Если диагноз трисомии поставлен до рождения ребенка, у родителей есть выбор — прервать беременность или родить неизлечимо больного ребенка. Семьи, которые считают прерывание беременности недопустимым, поддерживает Детский хоспис «Дом с маяком». Специалисты хосписа находятся рядом с родителями при родах и потом все время, пока паллиативный ребенок дома, сопровождают семью.
Хоспис обеспечивает такие семьи необходимым оборудованием, расходными материалами, лекарствами, специальным питанием, обучает родителей навыкам ухода за малышом. Кроме того, семью курируют врачи, психологи, социальные работники. Еще есть программа социальной передышки — можно оставить ребенка в стационаре на две недели в году, чтобы родители могли отдохнуть.
Первым ребенком, рожденным под опекой «Дома с маяком», была Маруся. Ее мама написала, что ей было очень сложно получить хоть какую-то информацию о том, какими рождаются дети с синдромом Эдвардса: «Я пересмотрела и перечитала терабайты историй западных семей с такими детьми, и они были очень светлыми, несмотря на весь мрак ситуации, я знала, что дети с синдромом Эдвардса бывают весьма крепкими, что совсем необязательно там будет страшный болевой синдром, судороги и асфиксия, что у них может быть короткая, но полная любви и заботы жизнь, однако как реализовать это в наших условиях, я не понимала. Результаты моих обследований говорили о том, что у Маруси были высокие шансы на благополучное рождение и хороший светлый промежуток».
Женщина обратилась в Детский хоспис. Потом она познакомилась с семьей Глаголевых, а Вероника Машкова рассказала ей, каким был ее сын Коля.
«Три месяца мы жили почти обычной жизнью. Маруся была очень славной, почти обычной девочкой. Мы начали гулять. Я строила планы лечения и реабилитации. Последний месяц был сложнее. За три дня произошло резкое ухудшение. Мне до сих пор сложно дается осознание того, что же все-таки произошло. Я не хотела верить, что наше время вышло, и здесь очень четко сработал врач «Дома с маяком»».

Маленький добрый гном…О мальчике, который прожил три месяца и изменил всех вокруг
Светлая печаль, как сама жизнь
Сын Вероники Машковой прожил три месяца. Коля тоже родился с синдромом Эдвардса. Вероника часто делится опытом переживания потери на встречах с родителями, потерявшими детей.
«Каждый раз возникает какое-то движение. Сначала приходит родитель, который вообще не может говорить, просто давится слезами, которые даже не текут. А со временем это живой, разговорчивый, общительный человек — да, плачущий, но это добрые слезы. Да, это потеря, это боль, ее стоит выплакать».
Вероника рассказывает, что сначала на этих встречах обычно просто молча слушала всех, кто пришел. Могла сказать что-то в конце, что отозвалось. Но часто поражалась: «Я не раз видела это чудо: родители приходили раздавленными, а поднимались распрямившимися и окрыленными, супруги приходили порознь, а уходили за руку…
Да, у нас тоже бывает печаль, но она светлая, как сама жизнь. Это ведь радость, что наши дети у нас были, и мы смогли это время вместе с ними прожить. А меня очень сильно поддерживало то, что все было не напрасно, что жизнь Коли и его история нужны людям».
У меня еще есть время, чтобы быть рядом
«Нам очень много помогал и помогает фонд «Вера», — рассказывает Екатерина Глаголева, — На момент, когда мы выписались первый раз из больницы, мы не знали ничего — что и как с Сашей делать. Фонд нас очень быстро «подхватил» — приехала медсестра, привезли кислородный концентратор, отсос, расходные материалы, нас научили, куда что вставлять. И стало не так страшно. Чувствовалась настоящая поддержка. Особенно актуальная на фоне округляющихся глаз врачей, которые наблюдают Сашу в районной поликлинике. Хотя у нас очень хороший участковый педиатр, но она сама признает, что таких детишек на участке больше нет, и она не знает, что делать в той или иной ситуации».

«Только молока прибавилось, как Тата от нас ушла»Какой ошибки удалось избежать и почему сейчас — не стыдно
И все же, чтобы принять диагноз Саши, родителям понадобилось больше года. Евгений Глаголев признается: «Ты оказываешься в ситуации, когда вообще не знаешь, что делать». Он уверен, что важно показать — вот, есть такие люди, кто через это прошел и справился, и готов делиться опытом, советом, помощью.
«Хочется сказать другим родителям, что первое ощущение, что у тебя нет опыта и в тебе только страх, потом пройдет. Это неизбежно. Это просто отсутствие информации. Как только она появится, сразу станет легче… Важно не забывать о себе. А родители всегда забывают о себе, они героически ухаживают за ребенком, но заканчивается это одинаково: без себя ты теряешь себя. И быстро. А потом уходит ощущение жизни. Ты в вечном подвиге, но подвиг вечным быть не может».
Родители Саши говорят, что состояние девочки постоянно очень тяжелое, у нее случаются боли неврологического характера, которые не купируются обычными анальгетиками. Иногда приступ удается снять только при помощи морфина. Но врачи часто считают, что детям такие лекарства противопоказаны. И однажды из-за того, что долго не снимался болевой синдром, у девочки произошел регресс состояния, начались приступы эпилепсии.
Но ребенок все равно ребенок, и Глаголевы замечают у дочери много забавного: «Наша Сашенька по характеру — настоящая маленькая женщина и это видно во всем ее поведении, манерах. Она очень смешно закрывает рукой глаза, это выглядит как «я устала, все свободны». Если она чего-нибудь не хочет, вы от нее не добьетесь этого».
Евгений Глаголев не задается вопросом, сколько времени отведено Саше: «Мы думали об этом, но это гадание на кофейной гуще. Если Саша уйдет — значит, наша жизнь будет какой-то другой».
Он написал в своем блоге:
«Мне близка мысль, что у каждого на этой земле своя миссия. И когда она закончена, тогда человек умирает. Неважно, молодой ты или старый: если все что мог/нужно было сделать — сделано, значит «пора-пора». Саша много раз могла умереть. Но сегодня мы проводим нашу девятую осень вместе. В сентябре всей семьей пережили ковид. Когда он нагрянул в 2020 году, внутри пробежало: «Неужели?!». Ведь очевидно, что Саша со своими больными легкими не перенесет его. Но нет. Улыбаюсь в очередной раз. И не могу не думать про только Ее миссию. Значит вот так. И у меня еще есть время, чтобы быть рядом. И любить здесь».
Вам может быть интересно:
 В класс пришел ребенок с неизлечимым заболеваниемУчитель русского языка и литературы дает советы коллегам, как сделать эту ситуацию комфортной для всех
В класс пришел ребенок с неизлечимым заболеваниемУчитель русского языка и литературы дает советы коллегам, как сделать эту ситуацию комфортной для всех  Когда ты четко понимаешь, что будет, а что — нет, проще становится житьИстория семьи, которая стала счастливой несмотря ни на что
Когда ты четко понимаешь, что будет, а что — нет, проще становится житьИстория семьи, которая стала счастливой несмотря ни на что  Как успеть все: подсказки родителям тяжелобольных детейМама двоих неизлечимо больных детей дает практические советы по управлению временем
Как успеть все: подсказки родителям тяжелобольных детейМама двоих неизлечимо больных детей дает практические советы по управлению временем  Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчасЛичная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчасЛичная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
Перепечатка материала в сети интернет возможна только при наличии активной гиперссылки на оригинал материала на сайте pro-palliativ.ru
Материал подготовлен с использованием гранта Президента Российской Федерации, предоставленного Фондом президентских грантов.
Использовано стоковое изображение от Depositphotos.
Что такое трисомия 18 и какие пороки развития она вызывает.
Дети с синдромом Эдвардса часто погибают еще в утробе матери, а если появляются на свет, редко доживают до трех месяцев. Виной всему — лишняя хромосома в 18-й паре. Но маленькому Ване из Белоруссии с этой хромосомной аномалией уже год и девять месяцев. Алёне из Тюмени — шесть лет. А самому старшему мальчику с этим синдромом уже шестнадцать.
Родители из сообщества «T18 Russia», объединяющего семьи с синдромом Эдвардса, рассказали, как сегодня диагностируют это заболевание. Как бы ни было трудно, эти мамы не сдались и своим примером доказывают, что такие ребята могут жить счастливо даже с тяжелым диагнозом.
Неуловимый диагноз
Инга ожидала своего третьего ребенка. На первом скрининге, когда обычно выявляют высокие риски рождения младенца с генетическими нарушениями (синдромы Дауна, Эдвардса, Патау, Тернера), отклонений в исследовании не обнаружили. По этой причине амниоцентез — дополнительное исследование с забором околоплодных вод, которое бы с высокой точностью показало наличие заболевания, проводить не стали. Инга благополучно доходила до 41-й недели и самостоятельно родила Ваню.
Момент, когда ей показали новорожденного, оказался непростым.
«Ты лежишь на родильном кресле и тебе показывают ребёнка, у которого нет части уха, закрыт глаз. Врачи говорят: мы не знаем, есть ли глаз вообще», — вспоминает женщина. У мальчика также обнаружили расщелину нёба и деформированные суставы пальцев. Врачи сразу предположили, что это синдром Пьера Робена — недоразвитие челюсти в сочетании с несращением мягкого нёба.

Особое строение челюсти не позволяет Ване дышать самостоятельно
«У большинства мам, которые состоят в родительском чате детей с синдром Эдвардса, во время беременности аномалия не диагностировалась. Это был «сюрприз» при рождении. У одних мам высокий риск рождения ребенка с аномалиями, как в моем случае — 1 к 33, определялся по крови, но показатели УЗИ были в норме. У других наоборот: кровь в норме, но визуализировались отклонения на УЗИ. Есть даже те, кто делал амниоцентез, но он не показал наличие синдрома. К сожалению, выявить эту патологию со 100% точностью не позволяет ни один метод диагностики», — говорит Наталья Евсеева, мама девочки с синдромом Эдвардса и организатор Сообщества родителей детей с синдромом Эдвардса России и стран СНГ «T18 Russia».
Диагноз найден
Новорожденного Ваню увезли в реанимацию и сразу подключили к аппарату искусственного дыхания. УЗИ мозга, сделанное в областном центре, показало полное отсутствие мозолистого тела — сплетения нервных волокон, которые должны соединять полушария мозга. В кардиохирургическом центре у Вани диагностировали порок сердца — двойное отхождение магистральных сосудов от правого желудочка, при котором вместо одной артерии в лёгкие идут две.
Около месяца маленький Ваня находился один в отделении реанимации на ИВЛ и зондовом питании. По недоразвитому «эльфийскому» уху, сложенным особым образом пальчикам на руках генетик предположила синдром Эдвардса. У мальчика взяли анализ на кариотип, и результат теста подтвердил трисомию 18-й хромосомы и полную форму синдрома.

Новорожденные с синдромом Эдвардса особым образом складывают пальцы
Синдром Эдвардса, или трисомия по 18-й хромосоме — это генетическое заболевание, при котором у ребенка образуется три хромосомы восемнадцатой пары вместо двух.
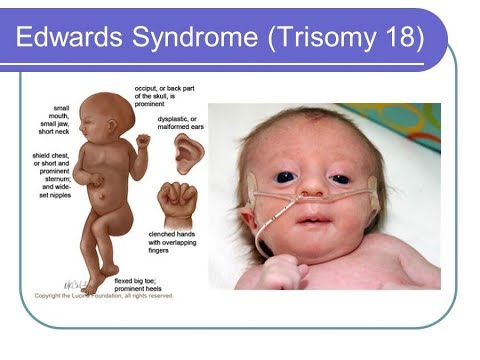
Дети с такой патологией рождаются с пониженной массой тела и множеством пороков развития: с измененной формой черепа, маленьким ртом, аномалиями твердого неба, узкими глазными щелями, искривленными ушными раковинами, дефектами конечностей, половых органов. Пальчики у них сжаты в кулак, но мизинец и указательный при этом располагаются поверх среднего и безымянного. Также синдром Эдвардса вызывает пороки сердца, часто не совместимые с жизнью и требующие хирургической коррекции.
Заболевание встречается у одного из 5000 новорожденных. На три-четыре больных девочки приходится один мальчик. Почти все беременности заканчиваются выкидышем. 40% детей с этим диагнозом не выживают во время родов. 60% детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10%.

Самым тяжелым испытанием в жизни Вани стала операция на сердце
«Сразу стало понятно, почему у ребенка столько пороков и чем они вызваны. Для нас этот диагноз был полной неожиданностью и вызвал шок, потому что наши старшие дети абсолютно здоровы. Но мы пережили тот тяжёлый период, когда у нас не было веры в ребёнка», — вспоминает Инга.
Мальчику сделали небольшую паллиативную операцию — установили клипсу на одну из артерий, чтобы уменьшить лёгочную гипертензию и дать ребёнку шанс выжить. Когда ему исполнился год и два месяца, его сердце прооперировали и полностью ликвидировали порок. После операции у ребенка были серьезные сбои сердечного ритма, обширная инфекция, угроза сепсис. Но Ваня выжил! Сейчас ему уже год и девять месяцев. Он дышит с помощью трахеостомы, питается через гастростому, но видит, слышит, может держать голову и даже начал проявлять эмоции.
«Пороки сердца — это наиболее часто встречающаяся патология у наших детей, — говорит Наталья Евсеева. — В большинстве своем они операбельные. У моей дочери Алёны размер отверстия в сердце был 15 мм, поэтому ей в три месяца проводили полостную операцию. В некоторых случаях дети с синдромом Эдвардса рождаются и без пороков сердца, но у них чаще всего присутствует дыхательная недостаточность. Если сердце плохо работает, нарушен кровоток, страдает головной мозг, соответственно идет отставание в физическом и умственном развитии, это тянет за собой цепочку тяжёлых осложнений, поэтому оперировать нужно всех детей — для них это огромный шанс жить и развиваться дальше. Среди детей с налаженной работой сердца есть те, которые разговаривают, изучают английский язык, ходят в детский сад и школу».
Поддержка своих
Сообщество родителей детей с синдромом Эдвардса России и стран СНГ «T18 Russia» существует уже четыре года. Сегодня в нем около 50 семей с детьми от 2 месяцев до 16 лет: диагноз у всех один, но генетические поломки различные, и состояние ребят тоже.
Организатором сообщества стала Наталья Евсеева из Тюмени — мама девочки Алёны с синдромом Эдвардса. Она родилась шесть лет назад с маленьким весом и пороком сердца. В три месяца ей сделали радикальную операцию на сердце, в два года еще две малоинвазивные операции. У девочки плохо работает кишечник, поэтому ей постоянно нужно принимать поддерживающую терапию. Слабые мышцы не позволяют самостоятельно ходить, ползать и сидеть, во всем необходима поддержка: родители используют вертикализатор, специальное домашнее кресло, на улице — коляску. Алёна узнает всех членов семьи, ярко выражает эмоции, произносит много звуков и слогов, но связно говорить не может.

Алёна дома, в своем специальном кресле с поддержкой спины и головы
Мария из Санкт-Петербурга, другая мама из сообщества «T18 Russia», рассказывает, что ждала здоровую крупную девочку, а родилась она маленькая и синяя. Сейчас Василисе уже 3 года и 7 месяцев. Ребенок чувствует себя хорошо, сидит практически самостоятельно, умеет стоять и делать шаги с поддержкой. Играет в игрушки, выбирает из них любимые. Знает родных людей, при незнакомых немного смущается, но легко идет на контакт, если видит позитивные эмоции.
«Развитие идет в очень замедленном темпе, сейчас Василиса развита примерно на 6-8 месяцев, — говорит Мария. — Но мы не унываем. За эти 3,5 года мы видели в больницах разных деток, которые в разы тяжелее Василиски. Моя девочка растет, развивается, общается, сама дышит и кушает! С ней можно поехать в путешествие, на экскурсию, даже на концерт, куда угодно! Мне кажется, сейчас у нас все хорошо».
У Надежды из Пскова пороки развития ребенка диагностировали после 30‑й недели беременности на УЗИ. Диагноз поставили уже после рождения дочери. Сегодня Еве три с половиной года, у нее неоперируемый порок сердца, белковая недостаточность и другие осложнения. Но при наличии всех проблем со здоровьем девочка сама дышит и ест с ложки. Ева пытается держать голову в специальном кресле, играет с игрушками, следит глазами за родителями и, кажется, все понимает на своём уровне. Из-за высокого легочного давления врачи федеральных центров пока не решаются оперировать сердце девочки. По словам мамы, самое трудное в жизни с особенной дочерью — вовремя получить качественную медицинскую помощь даже при обычной простуде: многие специалисты, узнав о синдроме, боятся назначать лекарства.

Ева с мамой Надеждой
Валентина из Краснодарского края рассказывает, что ей поставили высокий риск (1:17) рождения ребенка с синдром Эдвардса по крови на первом скрининге. Амниоцентез женщина делать отказалась, опасаясь тяжелых осложнений и боясь потерять ребенка. Сейчас ее мальчику уже четвертый год. Он самостоятельно ходит, все понимает, но не говорит. Саша посещает детский сад, занимается плаванием в бассейне. По словам мамы, больше всего трудностей вызывает кормление, так как ребенок пока не умеет жевать.
Есть в сообществе «T18 Russia» и русскоговорящие мамы, проживающие в других странах. Одна из них Алина из Цюриха. Ее дочке один год и 11 месяцев. Швейцарские врачи диагностировали у нее синдром Эдвардса на 12-13 неделе беременности с вероятностью 99%. Но родители приняли решение сохранить беременность. По словам мамы, сейчас малышка чувствует себя хорошо, пока не может держать голову, ползать и сидеть, но кушает с ложки, улыбается, любит путешествовать с семьей. «Самое трудное — неизвестность впереди. Очень хочется, чтобы она научилась сидеть, ходить и говорить. Но уверенности в этом нет, и это сложно принять, — рассказывает Алина, — но зато можно помогать ей, поддерживать, а главное безусловно любить,что мы и делаем всей семьей. В целом она как обычный малыш, даже спокойнее. Не капризничает без повода, радуется многому и дарит нам только положительные эмоции и много тепла. Она и есть настоящее чудо и свет. Порок сердца сгладился сам, а значит силы и ресурс у нее точно есть! Мы идем вперед, только маленькими шажками!»
Большинство детей сообщества «T18 Russia» дышат самостоятельно, едят с ложки и пьют из бутылочки или чашки. Их навыки самообслуживания — результат большой работы мам и специалистов. Как будет развиваться ребенок с частичной или полной трисомией 18-й хромосомы, предугадать нельзя. Все зависит от его внутренних резервов, заложенных природой, и поддержки семьи. Яркий тому пример — 16-летний мальчик с полной формой синдрома Эдвардса, который ходит в обычную школу, свободно разговаривает на английском языке и занимается плаванием.
«Наше сообщество — это локомотив, который помогает двигаться вперёд. Мы радуемся успехам детей, обсуждаем важные вопросы по здоровью, помогаем друг другу психологически и материально, делимся новой информацией о синдроме, подсказываем новым участникам, как правильно оформить инвалидность, получить технические средства реабилитации. К нам можно обратиться за любым советом и помощью», — говорит Наталья Евсеева.
Вступить в Сообщество родителей детей с синдромом Эдвардса России и стран СНГ «T18 Russia» и узнать больше о заболевании можно по ссылке.
Синдром Эдвардса — хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе.
Как часто встречается эта патология?
Дети с синдромом Эдвардса погибают на этапе внутриутробного развития приблизительно в 60% случаев. Несмотря на это, среди генетических заболеваний данный синдром у выживших младенцев является достаточно распространенным, по частоте встречаемости уступая только синдрому Дауна. Распространенность синдрома Эдвардса составляет 1 случай на 3 — 8 тысяч детей.
Считается, что у девочек данное заболевание встречается в 3 раза чаще, а риск синдрома Эдвардса значительно возрастает, если беременной женщине 30 и более лет.
Смертность при синдроме Эдвардса на первом году жизни составляет около 90%, причем средняя продолжительность жизни при тяжелом течении заболевания у мальчиков — 2–3 месяца, а девочек — 10 месяцев, а до взрослого состояния доживают лишь единицы. Чаще всего дети с синдромом Эдвардса умирают от удушья, пневмонии, сердечно–сосудистой недостаточности или кишечной непроходимости — осложнений, вызванных врожденными пороками развития.
Симптомы заболевания
Проявляется синдром у всех детей абсолютно по-разному: у некоторых возникают только основные симптомы заболевания, у других – проявляется их абсолютное большинство.
Различна у пациентов также и степень проявления имеющихся симптомов.
Итак, внешне синдром Эдвардса проявляется следующим образом:
- ребенок имеет маленькую голову по сравнению с туловищем;
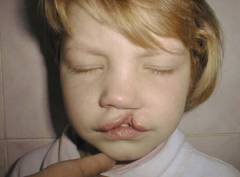
- узкие глаза (см. фото выше);
- плохо развитая нижняя челюсть;
- искажение черт и формы лица;
- удлиненные ушные раковины. Часто также отсутствуют некоторые составляющие части уха (мочка, слуховой канал);
- широкая и укороченная грудная клетка;
- верхняя губа совсем небольшого размера, маленький рот;
- сильно расширенная переносица, иногда «вдавленная» внутрь черепа;
- стопы ног неправильной формы;
- высокое небо с разрезом (волчья пасть);
- низкий лоб, затылок сильно выступает;
- косоглазие.
Помимо визуальных признаков, имеются и внутренние, куда более серьезные по своей природе:
- порок сердца;
- наличие грыж;
- отсутствие рефлексов, типичных для маленьких детей;
- недоразвитость мозжечка;
- удвоенные мочеточники; ;
- маленькая масса тела, около 2 кг при рождении;
- с возрастом развивается слабоумие;
- дистрофия мышц;
- онкология различных органов.
Синдром Эдвардса – диагностика
Описываемая генетическая болезнь является прямым показанием для прерывания беременности. Дети с синдромом Эдвардса никогда не смогут жить полноценно, а их здоровье будет стремительно ухудшаться. По этой причине важно диагностировать трисомию 18 на максимально раннем сроке. Для определения данной патологии разработано несколько информативных скринингов.
Анализ на синдром Эдвардса
Существуют неинвазивные и инвазивные методики исследования биологического материала. Второй тип тестов считается самым достоверным и надежным, он помогает выявить синдром Эдвардса у плода на ранних этапах развития. Неинвазивным является стандартный пренатальный скрининг крови матери. К инвазивным способам диагностики относятся:
- Биопсия ворсин хориона. Исследование проводится с 8 недели. Для осуществления анализа отщипывается кусочек оболочки плаценты, потому что его структура почти идеально совпадает с тканью плода.
- Амниоцентез. В ходе тестирования берется образец околоплодных вод. Эта процедура определяет синдром Эдвардса с 14 недели вынашивания.
- Кордоцентез. Для анализа требуется немного пуповинной крови плода, поэтому такой метод диагностики применяется исключительно на поздних сроках, с 20 недели.
Риск синдрома Эдвардса по биохимии
Пренатальный скрининг осуществляется в первом триместре беременности. Будущая мать должна сдать кровь в период с 11 по 13 неделю вынашивания для биохимического анализа. По результатам определения уровня хорионического гонадотропина и плазменного протеина А рассчитывается риск синдрома Эдвардса у плода. Если он высок, женщина вносится в соответствующую группу для следующего этапа исследований (инвазивных).
Синдром Эдвардса – признаки по УЗИ
Указанный вид диагностики используется редко, преимущественно в случаях, когда беременная не прошла предварительный генетический скрининг. Синдром Эдвардса на УЗИ можно выявить только на поздних сроках, когда плод уже почти полностью сформирован. Характерные симптомы трисомии 18:
- внутриутробные пороки сердечно-сосудистой и мочеполовой системы;
- аномалии опорно-двигательных структур;
- патологии костей черепа и мягких тканей головы.
- Косвенные признаки болезни на УЗИ:
- брадикардия;
- задержка развития плода;
- одна артерия в пуповине (должно быть две);
- грыжа в брюшной полости;
- визуальное отсутствие костей носа.
Лечение синдрома Эдвардса
Как и при других генетических заболеваниях, вызванных хромосомными аномалиями, прогноз для детей с синдромом Эдвардса неутешительный. Многие из них погибают сразу при рождении или в течение нескольких дней, несмотря на оказанную медицинскую помощь. Девочки могут прожить до десяти месяцев, мальчики погибают в течение первых двух-трех. Только 1% новорожденных доживает до десятилетнего возраста, при этом о самостоятельности и социальной адаптации не может быть и речи из-за серьезных нарушений интеллекта.
Больше шансов выжить в первые месяцы у пациентов с мозаичной формой синдрома, так как повреждения касаются не всех клеток организма. Мозаичная форма встречается в случае, если хромосомные аномалии произошли на этапе деления зиготы, уже после слияния мужской и женской половых клеток. Тогда та клетка, в которой было нерасхождение хромосом, из-за чего образовалась трисомия, при делении дает начало аномальным клеткам, что и провоцирует все патологические явления. Если же трисомия произошла на этапе гаметогенеза с одной из половых клеток, то аномальными будут все клетки плода.
Лекарственного средства, которое могло бы повысить шансы на выздоровление, не существует, так как пока не представляется возможным вмешательство на хромосомном уровне во всех клетках организма. Единственное, что может предложить современная медицина – симптоматическое лечение и поддержание жизнеспособности ребенка. Коррекция патологических явлений, связанных с синдромом Эдвардса, может улучшить качество жизни пациента, продлить ее срок. Хирургическое вмешательство при врожденных пороках развития нецелесообразно, так как влечет большие риски для жизни пациента и имеет множество осложнений.
Больные с синдромом Эдвардса с первых дней жизни должны наблюдаться у педиатра, так как они очень уязвимы для возбудителей инфекции. Среди новорожденных с данной патологией часто встречаются конъюнктивит, инфекционные заболевания мочеполовой системы, средний отит, синусит, пневмония.
Родителей ребенка с синдромом Эдвардса часто беспокоит вопрос – можно ли рожать еще раз, какова вероятность того, что следующая беременность также будет патологической. Исследования подтверждают, что риск повторного явления синдрома Эдвардса у одной и той же супружеской пары очень низок, даже в сравнении со средней вероятностью в 1% случаев. Вероятность рождение следующего ребенка с той же патологией составляет примерно 0,01%.
В целях своевременной диагностики синдрома Эдвардса будущим матерям рекомендуется проводить антенатальный скрининг в период беременности. При обнаружении патологий на ранних стадиях беременности можно будет сделать аборт по медицинским показаниям.
Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность — «Лечебное дело» в 1991 году, в 1993 году «Профессиональные болезни», в 1996 году «Терапия».
Наши авторы
Синдром Эдвардса: клинические проявления и возможности терапии

Синдром Эдвардса относят к хромосомным патологиям. Он характеризуется появлением дополнительной хромосомы в 18-ой паре. Синдром был описан в 1960 году Джоном Эдвардсом, поэтому и назван его именем. При развитии заболевания у больного отмечаются особенности внешнего вида: уменьшение размеров черепа, глаз и изменение формы ушей. Нарушается строение внутренних органов — сердца, сосудистой системы, органов мочевыделительной системы и пр. Выявить трисомию можно внутриутробно. Для этого существуют инвазивные и неинвазивные методы исследования. Эффективное лечение не разработано. Возможна только симптоматическая терапия, снижающая выраженность клинических признаков.
Причины развития
Основная причина развития заболевания — изменение хромосомного набора. Трисомия 18 встречается чаще всего. В некоторых случаях у пациентов обнаруживают мозаичный вариант патологии, сопровождающийся мутациями в конкретных генах.
Увеличение количества 18 хромосомы возникает в результате ее нерасхождения между клетками при мейозе (процесс деления половых клеток). Нарушения возникают в яйцеклетках. У ребенка развивается тяжелая форма заболевания. Легкие варианты болезни связаны с мозаицизмом. Это ситуация, при которой часть хромосомного материала переносится, приводя к нарушению работы генов.
В развитии хромосомных патологий важным факторов является возраст женщины. Если беременность возникла после 30 лет, то риск возникновения трисомии увеличивается в несколько раз.
Клинические проявления
Первые изменения при синдроме Эдвардса отмечаются в период беременности. При обследовании женщины выявляют многоводие, уменьшение размеров плаценты и наличие только одной пупочной артерии (в норме — 2 артерии). Масса детей при рождении меньше нормы — до 2100 г и менее, что связано с общей гипотрофией.
У детей с патологией выражены фенотипические изменения (особенности внешнего вида). При их наличии врач проводит дополнительное обследование для исключения синдрома Патау и других хромосомных аномалий. К характерным симптомам относят:
- долихоцефалические изменения формы черепа. Продольный размер головы преобладает над поперечным;
- уменьшение размера лба при выступающем затылке;
- у детей уменьшены глаза и рот.
Реже встречается кожная складка у внутреннего края глаза, опущение нижнего века, экзофтальм («выпученные» глаза), косоглазие и укорочение шеи. Деформация ушей представлена уменьшением мочек, отсутствием козелков и низким расположением ушных раковин.
Признаки заболевания включают изменения опорно-двигательного аппарата: деформацию пальцев кистей, уменьшение размеров грудины и ребер, врожденный вывих бедра, косолапость и пр. В связи с нарушением развития тканей у больных появляются папилломы и гемангиомы на кожном покрове.
18 хромосома у человека играет важную роль в формировании внутренних органов. Увеличение числа ее копий или транслокация (изменение расположения) участков приводит к аномалиям их развития. Наиболее часто встречаются дефекты сердечно-сосудистой системы в виде коарктации аорты, тетрады Фалло, изменения положения клапанов и др. Также страдает желудочно-кишечный тракт, мочеполовая система и центральная нервная система. Изменения в строении головного мозга характеризуются гидроцефалией, кистами, гипоплазией отдельных структур и т.п.
Синдром Эдвардса в большинстве случаев приводит к гибели пациентов в первый год жизни. Смерть возникает при отсутствии лечения из-за развития дыхательной или сердечно-сосудистой недостаточности.
Негативные последствия
Нарушения развития внутренних органов и центральной нервной системы приводит к множественным порокам. В связи с аномалиями строения сердца, главных сосудов и органов дыхания у ребенка развивается сердечно-сосудистая или дыхательная недостаточность. Она может стать причиной смерти при отсутствии своевременной медицинской помощи.
Пороки развития органов мочевыделения создают предпосылку для возникновения пиелонефрита, поликистозной почки и других аномалий. В результате их прогрессирования развивается хроническая почечная недостаточность, сопровождающаяся накоплением в крови продуктов азотистого обмена. Это может привести к нарушению работы ЦНС и другим тяжелым последствиям, включая летальный исход.
Последствиями микроцефалии и гипоплазии мозговых структур являются расстройства работы внутренних органов, а также невозможность формирования сложных психических или двигательных навыков. При поражении центров, ответственных за дыхание или сердечно-сосудистую деятельность, развивается их недостаточность.
Диагностика болезни
В диагностике заболевания важно ее внутриутробное выявление. При обнаружении болезни показано искусственное прерывание беременности. Косвенный признак патологии — недоразвитие одной из пупочных артерий, многоводие, уменьшение размеров плаценты и пр. Они могут быть выявлены во время проведения планового УЗИ.
Диагностической ценностью обладают неинвазивные методы, связанные с определением в крови матери маркеров болезни: хорионического гонадотропина, PAPP, альфа-фетопротеина, а также свободного эстрадиола. Изменение уровня указанных веществ в кровотоке женщины является показанием для проведения дополнительных диагностических процедур.
В процессе диагностики заболевания большое значение имеет совокупность факторов: результаты лабораторного скрининга и данных УЗИ, срок гестации и возраст беременной женщины. При наличии нескольких факторов рекомендованы инвазивные исследования — биопсия хориона, амниоцентез (взятие на анализ околоплодных вод) или кордоцентез (взятие анализа крови из пуповины). Полученный в результате процедур материал плода подвергается кариотипированию с подсчетом числа хромосом.
Больные после установления диагноза нуждаются в комплексном обследовании. Рекомендуется провести консультации невролога, кардиохирурга, кардиолога, уролога и других врачей. Всем детям проводят электрокардиографию, ЭхоКГ и ультразвуковое исследование внутренних органов. При наличии неврологического дефицита необходимо снятие ЭЭГ и УЗИ или КТ головного мозга.
Дифференциальную диагностику проводят с различными хромосомными патологиями — синдромом кошачьего крика и пр. В ее основе лежат молекулярно-генетические методы и определение кариотипа.
Подходы к лечению
Синдром Эдвардса заболевание хромосомное, с большим количеством пороков развития. Чаще всего дети с синдромом Эдвардса рождаются у женщин после 30 лет, девочек с этой патологией рождается в три раза больше, чем мальчиков.
Так как хромосомные аномалии не могут быть устранены, проводимое лечение направлено на поддержание жизни больного и устранение имеющихся симптомов. Терапия включает в себя медикаментозные и хирургические методики.
Лекарственные препараты назначаются в следующих случаях:
- наличие инфекционного поражения органов мочевыделительной или дыхательной системы. Применяют антибиотики широкого спектра действия, преимущественно из группы защищенных пенициллинов или цефалоспоринов;
- препараты, улучшающие мозговое кровообращение;
- средства, регулирующие тонус кровеносных сосудов.
Хирургические вмешательства выполняются по поводу аномалий развития сердца и сосудов, органов мочевыделительной системы, желудочно-кишечного тракта. Операции направлены на стабилизацию их состояния и улучшение качества жизни ребенка.
Прогноз для ребенка
Прогноз при хромосомных заболеваниях неблагоприятный. Выздоровление невозможно. Средняя продолжительность жизни — 1-2 года. До возраста 12 месяцев доживает менее 10% больных, а до 10 лет — только 1%. Для мозаичных вариантов синдрома Эдвардса прогноз благоприятнее, что связано с меньшей выраженности аномалий внутренних органов.
Основные причины гибели связаны с декомпенсацией сердечно-сосудистой и дыхательной недостаточности. Летальные состояния развиваются внезапно, несмотря на проводимое лечение.
Возможности профилактики
Профилактика болезни основывается на устранении причин, которые повышают риск развития хромосомных аномалий. К ним относят:
- беременность в возрасте до 30 лет. Чем возраст родителей выше, тем риск возникновения изменений в хромосомах больше;
- избегать работы на вредном химическом и промышленном производстве;
- исключение вредных привычек — табакокурения, употребления алкоголя и наркомании. Указанные воздействия в период беременности и после него способствуют накоплению в половых клетках мутаций, что может привести ко множеству хромосомных аномалий у будущего малыша, измененить его кариотип.
Важным элементом профилактики заболевания является скрининг беременных. Изменения, характерные для синдрома, хорошо видны на УЗИ. Назначение дополнительных инвазивных методик позволяет подтвердить диагноз и прервать беременность на любом сроке.
Развитие синдрома Эдвардса связано с изменениями в хромосомном наборе плода. Заболевание характеризуется неблагоприятным прогнозом, так как большинство детей умирает в первые годы жизни на фоне сердечно-сосудистой или дыхательной недостаточности. Лечение патологии носит симптоматический характер, так как исправление набора хромосом невозможно. Терапия направлена на устранение конкретных симптомов и увеличение продолжительности жизни больного ребенка. В процессе обследования важно исключить другие хромосомные аномалии — синдром Шерешевского-Тернера и пр. Это позволяет уточнить диагноз и прогноз для больного.
Видео
Читайте в следующей статье: синдром денди уокера
Комментарии
Узнавай и участвуй
Клубы на Бэби.ру — это кладезь полезной информации
Сайт предоставляет справочную информацию. Адекватная диагностика и лечение болезни возможны под наблюдением добросовестного врача. У любых препаратов есть противопоказания. Необходима консультация специалиста, а также подробное изучение инструкции!
 Синдром Эдвардса или трисомия 18 представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Синдром Эдвардса или трисомия 18 представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Распространенность синдрома Эдвардса по земному шару варьирует от 0,015 до 0,02%. Четкой зависимости от местности или расы не наблюдается. Статистически девочки болеют в 3 – 4 раза чаще мальчиков. Научного объяснения этой пропорции пока не выявлено. Тем не менее, отмечен ряд факторов, которые могут повысить риск возникновения этой патологии.
Как и другие хромосомные мутации, синдром Эдвардса является, в принципе, неизлечимым заболеванием. Самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка и способствовать определенному прогрессу в его развитии. Единых рекомендаций по уходу за такими детьми нет из-за огромного разнообразия возможных нарушений и осложнений.
Интересные факты
- Описание основных симптомов данной болезни было сделано еще в начале XX века.
- До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи.
- Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
- Реальная частота синдрома Эдвардса составляет 1 случай на 2,5 – 3 тысячи зачатий (0,03 – 0,04%), однако официальные данные значительно ниже. Это объясняется тем, что почти половина эмбрионов с данной аномалией не выживают и беременность заканчивается спонтанным абортом или внутриутробной смертью плода. Подробная диагностика причины выкидыша при этом проводится редко.
- Трисомия представляет собой вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе заболеваний. Помимо синдрома Эдвардса это синдромы Дауна (трисомия 21 хромосомы) и Патау (трисомия 13 хромосомы). При наличии других добавочных хромосом патология несовместима с жизнью. Лишь в этих трех случаях возможно рождение живого ребенка и его дальнейший (хоть и замедленный) рост и развитие.
Причины генетической патологии
 Синдром Эдвардса является генетическим заболеванием, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
Синдром Эдвардса является генетическим заболеванием, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
В каждой клетке человека имеется ядро, которое отвечает за хранение и обработку генетической информации. В ядре содержится 46 хромосом (23 пары), которые представляют собой многократно упакованную молекулу ДНК (дезоксирибонуклеиновая кислота). Эта молекула содержит в себе определенные участки, называемые генами. Каждый ген является прототипом определенного белка в организме человека. При необходимости клетка считывает информацию с этого прототипа и вырабатывает соответствующий белок. Дефекты генов ведут к производству аномальных белков, которые и ответственны за появление генетических заболеваний.
Хромосомная пара состоит из двух идентичных молекул ДНК (одна отцовская, другая – материнская), которые сцеплены между собой небольшим мостиком (центромерой). Место сцепления двух хромосом в паре предопределяет форму всего соединения и его вид под микроскопом.
Все хромосомы хранят различную генетическую информацию (о разных белках) и подразделяются на следующие группы:
- группа А включает 1 – 3 пару хромосом, которые отличаются большими размерами и X-образной формой;
- группа В включает 4 – 5 пару хромосом, которые также являются крупными, но центромера лежит дальше от центра, из-за чего форма напоминает букву Х со смещенным вниз или вверх центром;
- группа С включает 6 – 12 пару хромосом, которые по форме напоминают хромосомы группы В, но уступают им по размерам;
- группа D включает 13 – 15 пару хромосом, для которых характерны средние размеры и расположение центромеры у самого конца молекул, что придает сходство с буквой V;
- группа Е включает 16 – 18 пару хромосом, которые характеризуются маленькими размерами и срединным расположением центромеры (форма буквы Х);
- группа F включает 19 – 20 хромосомные пары, которые несколько мельче хромосом группы Е и схожи с ними по форме;
- группа G включает 21 – 22 пару хромосом, которые характеризуются V-образной формой и очень маленькими размерами.
Вышеперечисленные 22 пары хромосом называются соматическими или аутосомами. Кроме того существуют половые хромосомы, которые и составляют 23 пару. Они не похожи по внешнему виду, поэтому каждая из них обозначается отдельно. Женская половая хромосома обозначается Х и сходна с группой С. Мужская половая хромосома обозначается Y и сходна по форме и размерам с группой G. Если у ребенка обе хромосомы женские (тип ХХ), то рождается девочка. Если же одна из половых хромосом женская, а другая мужская, то рождается мальчик (тип ХY). Хромосомная формула называется кариотипом и может быть обозначена следующим образом — 46,ХХ. Здесь число 46 обозначает общее количество хромосом (23 пары), а ХХ – формулу половых хромосом, которая зависит от пола (в примере представлен кариотип нормальной женщины).
Синдром Эдвардса относится к так называемым хромосомным заболеваниям, когда проблема состоит не в дефекте гена, а в дефекте целой молекулы ДНК. Если быть более точным, то классическая форма этого заболевания подразумевает наличие лишней 18-й хромосомы. Кариотип в таких случаях обозначается как 47,ХХ, 18+ (для девочки) и 47,ХY, 18+ (для мальчика). Последняя цифра обозначает номер добавочной хромосомы. Излишек генетической информации в клетках приводит к появлению соответствующих проявлений болезни, которые и объединены под названием «синдром Эдвардса». Наличие дополнительной (третьей) хромосомы под номером 18 дало другое (более научное) название болезни – трисомия 18.
В зависимости от формы хромосомного дефекта различают три вида данного заболевания:
- Полная трисомия 18. Полная или классическая форма синдрома Эдвардса предполагает, что все клетки в организме имеют дополнительную хромосому. Данный вариант заболевания встречается более чем в 90% случаев и является наиболее тяжелым.
- Частичная трисомия 18. Частичная трисомия 18 является весьма редким феноменом (не более 3% от всех случаев синдрома Эдвардса). При ней в клетках организма содержится не целая дополнительная хромосома, а лишь ее фрагмент. Такой дефект может быть результатом неправильного деления генетического материала, но встречается он очень редко. Иногда часть восемнадцатой хромосомы присоединяется к другой молекуле ДНК (внедряется в ее структуру, удлиняя молекулу, или просто «цепляется» с помощью мостика). Последующее деление клеток приводит к тому, что в организме имеется 2 нормальные хромосомы номер 18 и еще часть генов с этих хромосом (сохранившийся фрагмент молекулы ДНК). В этом случае количество врожденных дефектов будет намного ниже. Наблюдается избыток не всей генетической информации, закодированной в 18-й хромосоме, а лишь ее части. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
- Мозаичная форма. Мозаичная форма синдрома Эдвардса встречается в 5 – 7% случаев данного заболевания. Механизм ее появления отличается от других видов. Дело в том, что здесь дефект образовался уже после слияния сперматозоида и яйцеклетки. Обе гаметы (половые клетки) изначально имели нормальный кариотип и несли по одной хромосоме каждого вида. После слияния сформировалась клетка с нормальной формулой 46,ХХ или 46,XY. В процессе деления этой клетки произошел сбой. При удвоении генетического материала один из фрагментов получил дополнительную 18-ю хромосому. Таким образом, на определенном этапе сформировался зародыш, часть клеток которого имеют нормальный кариотип (например, 46,ХХ), а часть – кариотип синдрома Эдвардса (47,ХХ, 18+). Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Форма получила название из-за того, что все клетки организма представляют собой своеобразную мозаику. Часть из них здорова, а часть – с тяжелой генетической патологией. Закономерности в распределении клеток в организме при этом не наблюдается, то есть все дефектные клетки не могут локализоваться только в одном месте, чтобы их можно было удалить. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
Наличие дополнительной хромосомы в геноме человека представляет множество проблем. Дело в том, что клетки человека запрограммированы считывать генетическую информацию и дублировать лишь заданное природой количество молекул ДНК. Нарушения даже в структуре одного гена могут привести к серьезным заболеваниям. При наличии же целой молекулы ДНК развиваются множественные нарушения еще на этапе внутриутробного развития до рождения ребенка.
Согласно последним исследованиям хромосома номер 18 содержит 557 генов, которые кодируют не менее 289 различных белков. В процентном отношении это примерно 2,5% всего генетического материала. Нарушения, которые вызывает столь большой дисбаланс, очень серьезны. Неправильное количество белков предопределяет множество аномалий в развитии различных органов и тканей. В случае синдрома Эдвардса чаще других страдают кости черепа, некоторые отделы нервной системы, сердечно-сосудистая и мочеполовая система. По всей видимости, это связано с тем, что гены, расположенные на этой хромосоме, имеют отношение к развитию именно этих органов и систем.
Таким образом, основной и единственной причиной синдрома Эдвардса является наличие дополнительной молекулы ДНК. Наиболее часто (при классической форме болезни) она наследуется от одного из родителей. В норме каждая гамета (сперматозоид и яйцеклетка) содержат по 22 непарные соматические хромосомы, плюс одна половая. Женщина всегда передает ребенку стандартный набор 22+Х, а мужчина может передать 22+Х либо 22+Y. Это предопределяет пол ребенка. Половые клетки родителей образуются в результате разделения обычных клеток на два набора. В норме материнская клетка делится на две равные части, но иногда не все хромосомы делятся пополам. Если 18-я пара не разошлась по полюсам клетки, то одна из яйцеклеток (или одни из сперматозоидов) будет заранее дефектным. В нем будет не 23, а 24 хромосомы. В случае если именно эта клетка будет участвовать в оплодотворении, ребенок получит дополнительную 18-ю хромосому.
На неправильное деление клеток могут повлиять следующие факторы:
- Возраст родителей. Доказано, что вероятность хромосомных аномалий увеличивается прямо пропорционально с возрастом матери. При синдроме Эдвардса эта связь менее выражена, чем при других похожих патологиях (например, синдром Дауна). Но для женщин старше 40 лет риск родить ребенка с данной патологией в среднем в 6 – 7 раз выше. Подобная зависимость от возраста отца наблюдается в значительно меньшей степени.
- Курение и алкоголь. Такие вредные привычки как курение и злоупотребление алкоголем могут действовать на половую систему человека, влияя на деление половых клеток. Таким образом, регулярное употребление этих веществ (а также других наркотических препаратов) повышает риск неправильного распределения генетического материала.
- Прием лекарственных средств. Некоторые лекарственные препараты при неправильном приеме в первом триместре могут повлиять на деление зародышевых клеток и спровоцировать мозаичную форму синдрома Эдвардса.
- Заболевания половой сферы. Перенесенные инфекции с поражением репродуктивных органов могут отразиться на правильном делении клеток. Они повышают риск хромосомных и генетических заболеваний в целом, хотя специально для синдрома Эдвардса подобные исследования не проводились.
- Радиационное излучение. Облучение половых органов рентгеновским излучением или другими ионизирующими излучениями может повлечь генетические мутации. Особенно опасно такое внешнее воздействие в подростковом возрасте, когда деление клеток происходит наиболее активно. Частицы, формирующие излучение, легко проникают сквозь ткани и подвергают молекулу ДНК своеобразной «бомбардировке». Если это происходит в момент деления клетки, риск хромосомной мутации особенно высок.
В целом же нельзя сказать, что причины развития синдрома Эдвардса окончательно известны и хорошо изучены. Вышеперечисленные факторы лишь повышают риск развития данной мутации. Не исключается и врожденная предрасположенность некоторых людей к неправильному распределению генетического материала в половых клетках. Например, считается, что у супружеской пары, которая уже родила ребенка с синдромом Эдвардса, вероятность появления второго ребенка с аналогичной патологией составляет аж 2 – 3% (примерно в 200 раз выше, чем средняя распространенность этой болезни).
Как выглядят новорожденные с синдромом Эдвардса?
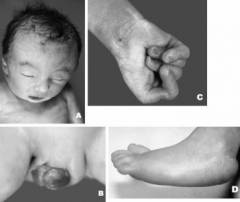 Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
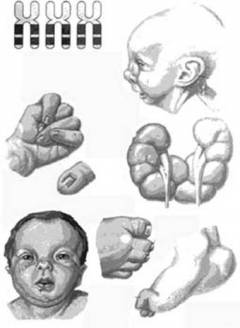
Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Изменение формы черепа
 Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Другим вариантом аномалии развития черепа является так называемая микроцефалия, при которой размеры головы в целом слишком малы по сравнению с остальным туловищем. Прежде всего, это касается не лицевого черепа (челюсти, скулы, глазницы), а именно черепной коробки, в которой располагается мозг. Микроцефалия менее характерна для синдрома Эдвардса, чем долихоцефалия, но она тоже встречается с более высокой частотой, чем среди здоровых людей.
Изменение формы ушной раковины
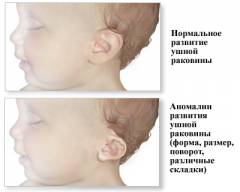 Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия). Сам слуховой проход обычно сужен, а примерно в 20 – 25% — вовсе отсутствует.
Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия). Сам слуховой проход обычно сужен, а примерно в 20 – 25% — вовсе отсутствует.
Аномалии развития неба
 Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком) у них идет продольная щель.
Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком) у них идет продольная щель.
Существует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой);
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти);
- полное незаращение твердого и мягкого неба;
- полное незаращение неба и губы.
В ряде случаев расщепление неба является двусторонним. Два выступающих вверх уголка верхней губы являются началом патологических щелей. Ребенок не может полностью закрыть рот из-за этого дефекта. В тяжелых случаях явно видно сообщение ротовой и носовой полости (даже при закрытом рте). Передние зубы в будущем могут отсутствовать или расти в сторону.
Данные дефекты развития известны также под названием волчья пасть, расщепление неба, заячья губа. Все они могут встречаться и не в рамках синдрома Эдвардса, однако у детей с этой патологией их частота особенно высока (почти 20% новорожденных). Значительно чаще (до 65% новорожденных) обладают другой особенностью, известной как высокое или готическое небо. Оно может быть отнесено к вариантам нормы, так как встречается и у здоровых людей.
Наличие расщепленного верхнего неба или верхней губы еще не подтверждает синдром Эдвардса. Этот порок развития может встречаться с довольно высокой частотой и самостоятельно без сопутствующих нарушений со стороны других органов и систем. Для исправления данной аномалии существует ряд стандартных хирургических вмешательств.
Стопа-качалка
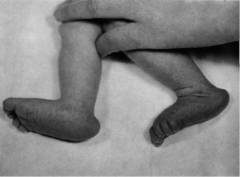 Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Внешне стопа у новорожденного ребенка выглядит следующим образом. Пяточный бугор, на который опирается задняя часть стопы, выдается назад. Свод при этом может полностью отсутствовать. Это легко заметить, посмотрев на стопу с внутренней стороны. В норме там вырисовывается вогнутая линия, направляющаяся от пятки к основанию большого пальца. При стопе-качалке этой линии нет. Стопа плоская или даже выпуклая. Это и придает ей сходство с ножками кресла-качалки.
Аномальная длина пальцев
 У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У взрослых укорочение большого пальца на ноге не имеет такой диагностической ценности. Подобный дефект может быть индивидуальной особенностью у здорового человека или следствием воздействия других факторов (деформация суставов, заболевания костей, ношение обуви, не соответствующей по размеру). В связи с этим данный признак нужно рассматривать как возможный симптом только у новорожденных детей при наличии других аномалий развития.
Изменение формы нижней челюсти
 Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
У новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны);
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Зазор между нижней и верхней челюстью может составлять более 1 см, что очень много, учитывая размеры головы малыша.
Сращение пальцев
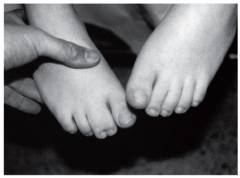 Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Синдактилия встречается не только при синдроме Эдвардса, но и при многих других хромосомных заболеваниях. Известны и случаи, когда этот порок развития являлся единственным, и в остальном больной ничем не отличался от нормальных детей. В связи с этим сращение пальцев является лишь одним из возможных признаков синдрома Эдвардса, который помогает заподозрить диагноз, но не подтверждает его.
Аномалии развития половых органов
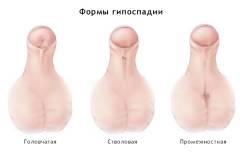 Непосредственно после родов у новорожденных с синдромом Эдвардса иногда можно наблюдать аномалии развития внешних половых органов. Как правило, они сочетаются с дефектами развития всего мочеполового аппарата, однако без специальных диагностических мероприятий это установить невозможно. Наиболее же частыми аномалиями, заметными внешне, являются недоразвитие полового члена у мальчиков и гипертрофия (увеличение в размерах) клитора у девочек. Они встречаются примерно в 15 – 20% случаев. Несколько реже может наблюдаться аномальное расположение мочеиспускательного канала (гипоспадия) или отсутствие яичек в мошонке у мальчиков (крипторхизм).
Непосредственно после родов у новорожденных с синдромом Эдвардса иногда можно наблюдать аномалии развития внешних половых органов. Как правило, они сочетаются с дефектами развития всего мочеполового аппарата, однако без специальных диагностических мероприятий это установить невозможно. Наиболее же частыми аномалиями, заметными внешне, являются недоразвитие полового члена у мальчиков и гипертрофия (увеличение в размерах) клитора у девочек. Они встречаются примерно в 15 – 20% случаев. Несколько реже может наблюдаться аномальное расположение мочеиспускательного канала (гипоспадия) или отсутствие яичек в мошонке у мальчиков (крипторхизм).
Флексорное положение кистей
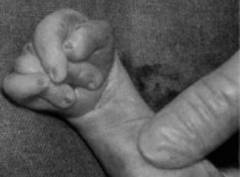 Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Дерматоглифические признаки
 При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
Основными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
Специальные исследования могут выявить и другие отклонения от нормы, однако непосредственно после рождения, без привлечения узких специалистов, врачам достаточно этих изменений.
Помимо вышеперечисленных признаков существует еще целый ряд возможных аномалий развития, которые могут помочь в предварительной диагностике синдрома Эдвардса. По некоторым данными при подробном внешнем осмотре можно обнаружить до 50 внешних признаков. Сочетание наиболее частых симптомов, представленных выше, с высокой вероятностью говорит о наличии у ребенка этой тяжелой патологии. При мозаичном варианте синдрома Эдвардса множественных аномалий может и не быть, однако наличие даже одного из них является показанием к проведению специального генетического теста.
Как выглядят дети с синдромом Эдвардса?
 У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
Большинство внешних проявлений синдрома, замеченных при рождении, остаются и становятся более заметными. Речь идет о форме черепа, стопе-качалке, деформации ушной раковины и т. п. Постепенно к ним начинают прибавляться и другие внешние проявления, которые невозможно было заметить сразу после рождения. В данном случае речь идет о признаках, которые могут появиться у детей в первый год жизни.
Дети с синдромом Эдвардса имеют следующие внешние особенности:
- отставание в физическом развитии;
- косолапость;
- аномальный тонус мышц;
- ненормальные эмоциональные реакции.
Отставание в физическом развитии
Отставание в физическом развитии объясняется низкой массой тела ребенка при рождении (всего 2000 – 2200 г при нормальном сроке беременности). Значительную роль играет и генетический дефект, который не позволяет всем системам организма нормально и гармонично развиваться. Основные показатели, по которым оценивают рост и развитие ребенка, сильно снижены.
Заметить отставание ребенка можно по следующим антропометрическим показателям:
- рост ребенка;
- вес ребенка;
- окружность грудной клетки;
- окружность головы (данный показатель может быть в норме или даже увеличен, но на него нельзя полагаться из-за врожденной деформации черепа).
Косолапость
Косолапость является следствием деформации костей и суставов стоп, а также отсутствия нормального контроля со стороны нервной системы. Дети с трудом начинают ходить (большинство не доживает до этого этапа из-за врожденных пороков развития). Внешне о наличии косолапости можно судить по деформации стоп, ненормальному положению ног в состоянии покоя.
Аномальный тонус мышц
Аномальный тонус, который при рождении вызывает флексорное положение кисти, по мере роста начинает проявляться и на других группах мышц. Чаще всего у детей с синдромом Эдвардса сила мышц снижена, они вялые и лишены нормального тонуса. В зависимости от характера повреждений центральной нервной системы некоторые группы могут иметь повышенный тонус, что проявляется спастическими сокращениями этих мышц (например, сгибатели рук или разгибатели ног). Внешне это проявляется отсутствием минимальной координации движений. Иногда спастические сокращения ведут к ненормальным перегибам конечностей или даже к вывихам.
Ненормальные эмоциональные реакции
Отсутствие или ненормальное проявление каких-либо эмоций является следствием аномалий в развитии некоторых отделов мозга (чаще всего мозжечка и мозолистого тела). Эти изменения приводят к серьезному отставанию в умственном развитии, которое наблюдается у всех без исключения детей с синдромом Эдвардса. Внешне низкий уровень развития проявляется характерным «отсутствующим» выражением лица, отсутствием эмоционального ответа на внешние раздражители. Ребенок плохо поддерживает визуальный контакт (не следит за движущимся перед глазами пальцем и т. п.). Отсутствие реакции на резкие звуки может быть следствием поражения как нервной системы, так и слухового аппарата. Все эти признаки обнаруживаются по мере роста ребенка в первые месяцы жизни.
Как выглядят взрослые с синдромом Эдвардса?
 В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
При более легкой мозаичной форме, когда лишь часть клеток в организме содержит аномальный набор хромосом, выживаемость несколько больше. Однако даже в этих случаях до взрослого возраста доживают единичные пациенты. Их внешний вид определяется врожденными аномалиями, которые присутствовали при рождении (заячья губа, деформированная ушная раковина, и др.). Основным же симптомом, присутствующим у всех без исключения детей, является серьезнейшее отставание в умственном развитии. Дожив до взрослого возраста, ребенок с синдромом Эдвардса является глубоким олигофреном (IQ менее 20, что соответствует самой тяжелой степени умственной отсталости). В целом же в медицинской литературе описываются единичные случаи, когда дети с синдромом Эдвардса доживали до совершеннолетнего возраста. Из-за этого накоплено слишком мало объективных данных, чтобы говорить о внешних признаках этого заболевания у взрослых.
Диагностика генетической патологии
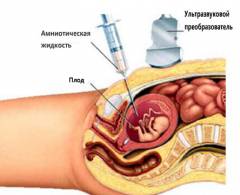 В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
Диагностика синдрома Эдвардса возможна на следующих этапах:
- диагностика до момента зачатия;
- диагностика во время внутриутробного развития;
- диагностика после рождения.
Диагностика до момента зачатия
Диагностика до момента зачатия ребенка является идеальным вариантом, но, к сожалению, на современном этапе развития медицины ее возможности очень ограничены. Врачи могут с помощью нескольких методов предположить повышенную вероятность рождения ребенка с хромосомным заболеванием, но не более того. Дело в том, что при синдроме Эдвардса, в принципе, нарушения у родителей обнаружить нельзя. Дефектная половая клетка с 24 хромосомами является лишь одной из многих тысяч. Поэтому сказать наверняка до момента зачатия, родится ли ребенок с данным заболеванием, нельзя.
Основными методами диагностики до момента зачатия являются:
- Семейный анамнез. Семейный анамнез представляет собой подробный опрос обоих родителей об их родословной. Врача интересуют любые случаи наследственных (и особенно хромосомных) заболеваний в семье. Если хотя бы один из родителей припоминает случай трисомии (синдром Эдвардса, Дауна, Патау), это сильно повышает вероятность рождения больного ребенка. Однако риск все равно составляет не более 1%. При повторных случаях этих заболеваний у предков риск многократно возрастает. По сути, анализ сводится к консультации у неонатолога или генетика. Предварительно родители могут постараться собрать более подробную информацию о своих предках (желательно на 3 – 4 колена). Это повысит точность данного метода.
- Обнаружение факторов риска. Основным фактором риска, который объективно повышает риск хромосомных аномалий, является возраст матери. Как уже говорилось выше, у матерей после 40 лет вероятность рождения ребенка с синдромом Эдвардса многократно возрастает. По некоторым данным, после 45 лет (возраст матери) почти каждая пятая беременность сопровождается хромосомной патологией. Большинство из них заканчивается выкидышем. Другими факторами являются перенесенные инфекционные заболевания, хронические болезни, вредные привычки. Однако их роль в диагностике значительно более низкая. Точного ответа на вопрос, будет ли зачат ребенок с синдромом Эдвардса, этот метод тоже не дает.
- Генетический анализ родителей. Если предыдущие методы сводились к опросу родителей, то генетический анализ представляет собой полноценное исследование, которое требует наличия специальной аппаратуры, реактивов и квалифицированных специалистов. У родителей берется кровь, из которой в лаборатории выделяют лейкоциты. После обработки специальными веществами в этих клетках становятся хорошо видны хромосомы на стадии деления. Таким образом составляется кариотип родителей. В подавляющем большинстве случаев он нормальный (при хромосомных нарушениях, которые могут быть здесь обнаружены, вероятность продолжения рода ничтожно мала). Кроме того с помощью специальных маркеров (фрагменты молекулярных цепочек) можно обнаружить участки ДНК с дефектными генами. Однако здесь будут обнаружены не хромосомные нарушения, а генетические мутации, которые не влияют напрямую на вероятность синдрома Эдвардса. Таким образом, генетический анализ родителей до момента зачатия, несмотря на сложность и высокую стоимость, также не дает однозначного ответа относительно прогнозов на данную патологию.
Диагностика во время внутриутробного развития
В период внутриутробного развития существует несколько способов, которые могут прямо или косвенно подтвердить наличие у зародыша хромосомной патологии. Точность этих методов значительно выше, так как врачи имеют дело не с родителями, а с самим организмом плода. Для изучения доступен как сам зародыш, так и его клетки с собственным ДНК. Данный этап называется также пренатальной диагностикой и является наиболее важным. В это время можно подтвердить диагноз, предупредить родителей о наличии патологии и при необходимости прервать беременность. Если же женщина решит рожать и новорожденный будет жив, то врачи получат возможность заранее подготовиться к оказанию ему необходимой помощи.
Основными методами исследования в рамках пренатальной диагностики являются:
- Ультразвуковое исследование (УЗИ). Данный метод является неинвазивным, то есть не предполагает повреждения тканей матери или плода. Он полностью безопасен и рекомендован для всех беременных женщин в рамках пренатальной диагностики (независимо от их возраста или повышенного риска для хромосомных заболеваний). Стандартная программа предполагает, что УЗИ надо делать трижды (на 10 – 14, 20 – 24 и 32 – 34 неделе беременности). Если лечащий врач предполагает возможность врожденных аномалий развития, могут быть проведены и незапланированные УЗИ. О синдроме Эдвардса может говорить отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития (микроцефалия, деформация костей). Эти нарушения с высокой вероятностью говорят о тяжелых генетических заболеваниях, но синдром Эдвардса окончательно подтвердить все же невозможно.
- Амниоцентез. Амниоцентез представляет собой цитологический (клеточный) анализ околоплодных вод. Врач аккуратно вводит специальную иглу под контролем аппарата УЗИ. Прокол делается в месте, где нет петель пупочного канатика. С помощью шприца берется необходимое для исследования количество амниотической жидкости. Процедуру можно проводить во всех триместрах беременности, но оптимальным сроком для диагностики хромосомных нарушений является период после 15 недели беременности. Частота осложнений (вплоть до спонтанного прерывания беременности) составляет до 1%, поэтому процедуру не стоит проводить при отсутствии каких-либо показаний. После забора околоплодных вод проводится обработка полученного материала. В них жидкости имеются клетки с поверхности кожи малыша, которые содержат образцы его ДНК. Именно их и проверяют на наличие генетических заболеваний.
- Кордоцентез. Кордоцентез представляет собой наиболее информативный метод пренатальной диагностики. После обезболивания и под контролем аппарата УЗИ врач прокалывает с помощью специальной иглы сосуд, проходящий в пуповине. Таким образом, получают образец крови (до 5 мл) развивающегося ребенка. Техника выполнения анализа аналогична таковой для взрослых. Данный материал можно с высокой точностью исследовать на предмет различных генетических аномалий. В том числе можно сделать кариотипирование плода. При наличии дополнительной 18-й хромосомы можно говорить о подтвержденном синдроме Эдвардса. Данный анализ рекомендуется проводить после 18-й недели беременности (оптимально 22 – 25 недели). Частота возможных осложнений после кордоцентеза составляет 1,5 – 2%.
- Биопсия хориона. Хорион представляет собой одну из зародышевых оболочек, содержащую клетки с генетической информацией плода. Данное исследование предполагает пункцию матки под наркозом через переднюю брюшную стенку. С помощью специальных биопсийных щипцов берут образец ткани для анализа. Затем проводится стандартное генетическое исследование полученного материала. Для диагностики синдрома Эдвардса делается кариотипирование. Оптимальным сроком для проведения биопсии хориона считают 9 – 12 неделю беременности. Частота осложнений составляет 2 – 3%. Основным преимуществом, отличающим его от других методов, является скорость получения результата (уже через 2 – 4 дня).
Диагностика после рождения
Диагностика синдрома Эдвардса после рождения является наиболее легкой, быстрой и точной. К сожалению, на этот момент уже произошло появление на свет ребенка с тяжелой генетической патологией, эффективного лечения которой в наше время пока не существует. Если на этапе пренатальной диагностики заболевание не было выявлено (либо соответствующие исследования не проводились), то подозрение на синдром Эдвардса появляется сразу после рождения. Ребенок обычно доношен или даже переношен, но его масса все равно ниже среднего показателя. Кроме того, обращают на себя внимание некоторые врожденные дефекты, о которых говорилось выше. Если их замечают, проводят генетический анализ для подтверждения диагноза. У ребенка берется кровь для анализа. Однако на данном этапе подтвердить наличие синдрома Эдвардса – неглавная проблема.
Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения.
Для обнаружения пороков в развитии внутренних органов применяют следующие методы исследования:
- ультразвуковое исследование брюшной полости;
- эхокардиография;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи;
- компьютерная томография или магнитно-резонансная томография;
- рентгенография.
Для рентгеновского обследования младенческий возраст обычно считается противопоказанием, но в данном случае опасностью можно пренебречь. Дело в том, что речь идет об обнаружении патологий, которые представляют опасность для жизни непосредственно в данный момент. Поэтому врачи должны срочно получить всю необходимую информацию об имеющихся пороках развития. Это поможет выбрать тактику лечения. В большинстве случаев грамотное ведение пациента и соответствующий уход помогают продлить жизнь ребенка.
Таким образом, можно заключить, что существует много методов диагностики синдрома Эдвардса. Некоторые из них (амниоцентез, кордоцентез и др.) представляют определенную опасность осложнений и не проводятся без специальных показаний. Основными показаниями считается наличие в роду случаев хромосомных заболеваний и возраст матери старше 35 лет. Программа диагностики и ведения пациентки на всех этапах беременности может быть изменена лечащим врачом по необходимости.
Прогноз для детей с синдромом Эдвардса
 Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований) говорят, что больше половины детей (50 – 55%) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований) говорят, что больше половины детей (50 – 55%) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
При частичной трисомии или мозаичной форме синдрома прогноз несколько лучше. Средняя продолжительность жизни при этом увеличивается до нескольких лет. Это объясняется тем, то аномалии развития при более легких формах не ведут так быстро к смерти ребенка. Тем не менее, основная проблема, а именно серьезное отставание в умственном развитии, присуща всем без исключения больным. При достижении подросткового возраста нет шансов ни на продолжение потомства (половая зрелость обычно не наступает), ни на возможность работы (даже механической, не требующей особых навыков). Существуют специальные центры для ухода за детьми с врожденными заболеваниями, где больным с синдромом Эдвардса обеспечивают уход и по возможности способствуют их интеллектуальному развитию. При достаточных усилиях со стороны врачей и родителей ребенок, проживший больше года, может научиться улыбаться, реагировать на движение, самостоятельно поддерживать положение тела или питаться (при отсутствии пороков системы пищеварения). Таким образом, признаки развития все же наблюдаются.
Высокая детская смертность при данном заболевании объясняется большим количеством пороков развития внутренних органов. Они незаметны непосредственно при рождении, но присутствуют практически у всех больных. В первые месяцы жизни дети обычно умирают от остановки сердца или дыхания.
Чаще всего пороки развития наблюдаются в следующих органах и системах:
- опорно-двигательный аппарат (кости и суставы, включая череп);
- сердечно-сосудистая система;
- центральная нервная система;
- пищеварительная система;
- мочеполовая система;
- другие нарушения.
Опорно-двигательный аппарат
Основными пороками в развитии опорно-двигательного аппарата является аномальное положение пальцев и искривление стоп. В бедренном суставе наблюдается сведение ног таким образом, что колени почти соприкасаются, а стопы смотрят немного в стороны. Нередко у детей с синдромом Эдвардса обнаруживается необычно короткая грудина. Это деформирует грудную клетку в целом и создает проблемы с дыханием, которые усугубляются по мере роста, даже если сами легкие не затронуты.
Дефекты развития черепа являются в основном косметическими. Однако такие пороки как волчья пасть, заячья губа и высокое небо создают серьезные трудности с кормлением ребенка. Нередко до проведения операций по исправлению этих дефектов ребенка переводят на парентеральное питание (в виде капельниц с питательными растворами). Другим вариантом является использование гастростомы – специального зонда, через который пища попадает прямо в желудок. Его установление требует отдельного хирургического вмешательства.
В целом пороки развития опорно-двигательного аппарата не создают прямой угрозы для жизни ребенка. Однако косвенно они влияют на его рост и развитие. Частота таких изменений у больных синдромом Эдвардса составляет около 98%.
Сердечно-сосудистая система
Пороки развития сердечно-сосудистой системы являются основной причиной смерти в раннем детском возрасте. Дело в том, что подобные нарушения встречаются почти в 90% случаев. Чаще всего они серьезно нарушают процесс транспорта крови по организму, приводя к выраженной сердечной недостаточности. Большинство сердечных патологий может быть исправлено хирургическим путем, но не каждому ребенку можно провести такую сложную операцию.
Наиболее частыми аномалиями со стороны сердечно-сосудистой системы являются:
- незаращение межпредсердной перегородки;
- незаращение межжелудочковой перегородки;
- сращение створок клапанов (или, наоборот, их недоразвитие);
- коарктация (сужение) аорты.
Все эти пороки сердца ведут к серьезным нарушениям кровообращения. Артериальная кровь не поступает в нужном объеме к тканям, из-за чего клетки организма начинают гибнуть.
Центральная нервная система
Самым характерным пороком со стороны центральной нервной системы является недоразвитие мозолистого тела и мозжечка. Это причина самых разнообразных нарушений, в том числе и умственной отсталости, которая наблюдается у 100% детей. Кроме того, нарушения на уровне головного и спинного мозга вызывают аномальный тонус мышц и предрасположенность к судорогам или спастическим сокращениям мышц.
Пищеварительная система
Частота пороков пищеварительной системы при синдроме Эдвардса составляет до 55%. Чаще всего эти аномалии развития представляют серьезную угрозу жизни ребенка, поскольку не позволяют ему нормально усваивать питательные вещества. Питание же в обход естественных органов пищеварения сильно ослабляет организм и усугубляет состояние ребенка.
Наиболее частыми пороками развития со стороны пищеварительной системы являются:
- дивертикул Меккеля (слепой отросток в тонкой кишке);
- атрезия пищевода (зарастание его просвета, из-за чего пища не проходит в желудок);
- атрезия желчных путей (накопление желчи в пузыре).
Все эти патологии требуют хирургического исправления. В большинстве случаев операция помогает лишь немного продлить жизнь ребенку.
Мочеполовая система
Наиболее серьезные пороки со стороны мочеполовой системы связаны с нарушением работы почек. В ряде случаев наблюдается атрезия мочеточников. Почка с одной стороны может быть дублирована или сращена с лежащими рядом тканями. Если имеет место нарушение фильтрации, в организме со временем начинают накапливаться токсичные продукты жизнедеятельности. Кроме того может наблюдаться рост артериального давления и нарушения в работе сердца. Серьезные аномалии развития почек представляют прямую угрозу для жизни.
Другие нарушения
Другими возможными нарушениями развития являются грыжи (пупочная, паховая). Могут обнаруживаться и дисковые грыжи позвоночника, которые приведут к неврологическим проблемам. Со стороны глаз иногда наблюдается микрофтальмия (маленький размер глазных яблок).
Совокупность этих пороков развития предопределяет высокую детскую смертность. В большинстве случаев, если синдром Эдвардса диагностирован на ранних этапах беременности, врачи рекомендуют делать аборт по медицинским показаниям. Тем не менее, окончательное решение принимает сама пациентка. Несмотря на всю серьезность заболевания и плохой прогноз многие предпочитают надеяться на лучшее. Но, к сожалению, в ближайшее время серьезных сдвигов в методах диагностики и лечения синдрома Эдвардса, судя по всему, не предвидится.