Индикаторные погрешности титрования
При титровании возможны случайные и
систематические погрешности. Случайные
погрешности связаны с измерением объема
и массы навески, но и значительную часть
погрешности титрования составляют
систематические погрешности, в частности,
индикаторная.
Случайные погрешности обрабатываются
по законам математической статистики.
Индикаторные погрешности связаны с
тем, что pT индикатора не
совпадает со значением pH
в ТЭ. Конечная точка титрования с данным
индикатором не совпадает с ТЭ.
При недотитровывании:
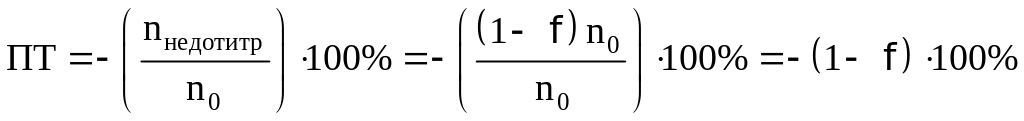
При перетитровывании:
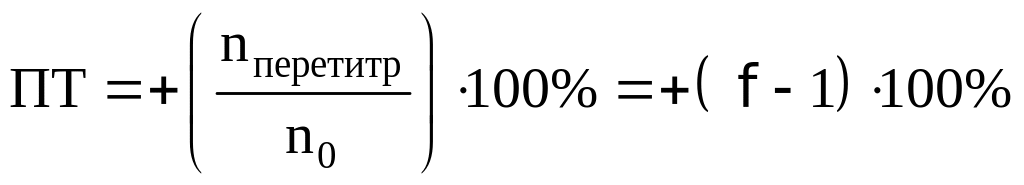
Возьмем индикаторы хризоидин (pT=5,50)
и хлорфеноловый красный (pT
= 5,80). В данном случае в КТТ pH
будет больше, чем pH в ТЭ
(pH = 5,28), а, следовательно,
в растворе будет неотитрованное
основание. Эта погрешность, обусловленная
содержанием неоттитрованного основания,
называется щелочной, будет определяться
уравнением
![]()
.
На данном этапе титрования pH
будет определяться по формуле
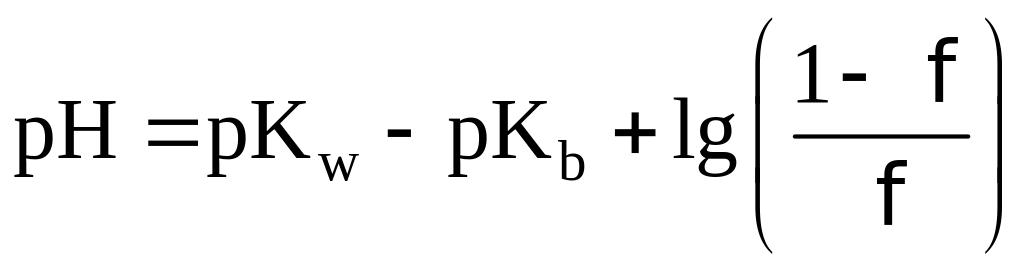
.
В КТТ pH раствора равен
pT. Следовательно, можно
найти и f
в КТТ:
![]()
.
Отсюда,

.
Посчитаем ПТ для данных индикаторов:
хризоидин:
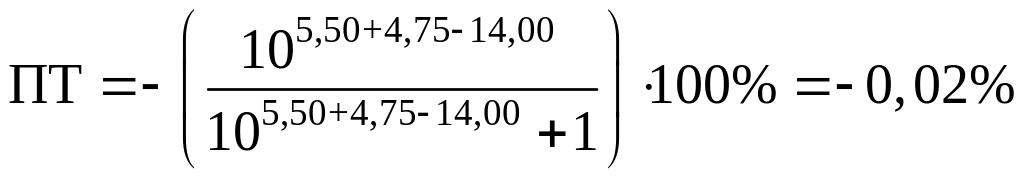
хлорфеноловый красный:
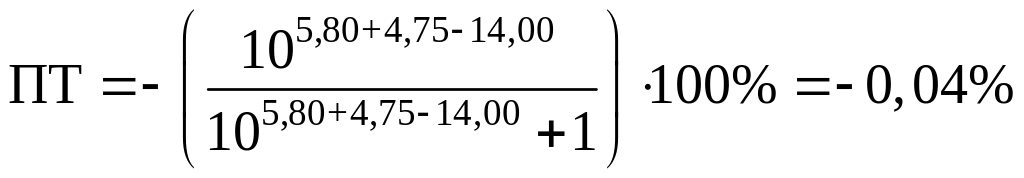
Оба эти индикатора подойдут для нашего
титрования. Рассмотрим ПТ для индикатора
розоловая кислота (pT=7,1).
По данной формуле получается ПТ=–0,70%,
что превышает обычно задаваемое значение
погрешности (±0,2%).
Рассмотрим же теперь случай, когда мы
используем индикаторы с pT
меньшим, чем pH в ТЭ. В КТТ
раствор будет перетитрован, и pH
будет определяться концентрацией
сильной кислоты (водородная погрешность),
и в нашей задаче определяться уравнением
.
Погрешность будет определяться по
формуле
![]()
Возьмем для примера индикаторы лакмоид
(pT=5,20), ализариновый красный
C (pT=4,45) и
бромфеноловый синий (pT=3,80).
pH раствора в КТТ равен
pT:
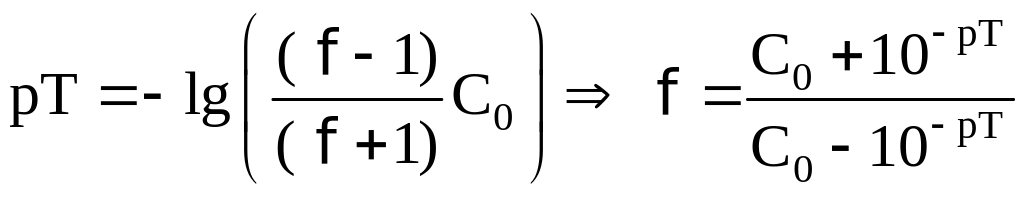
.
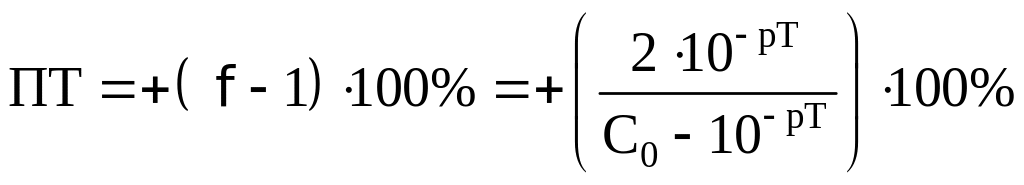
.
Рассчитаем ПТ для наших индикаторов:
Лакмоид:

Ализариновый красный C:
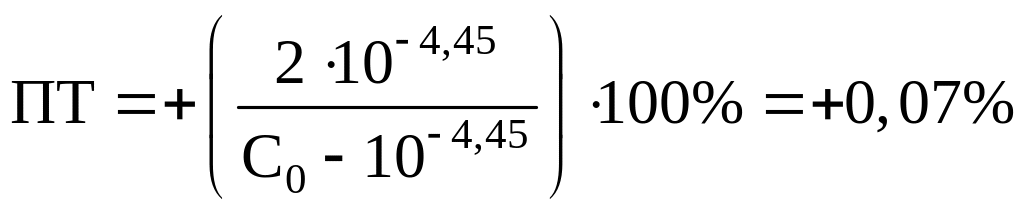
Бромфеноловый синий:

Вполне очевидно, что из двух предложенных
индикаторов наиболее подходящим является
ализариновый красный C.
Учитывая все расчеты, приходим к выводу,
что самыми подходящими для нашего опыта
индикаторами являются хризоидин (с
интервалом перехода 4,0 – 7,0, оранжевая
– желтая) и лакмоид (4,0 – 6,4, красная –
синяя).
Выводы
По кривой титрования аммиака можно
сделать ряд выводов.
В ходе титрования заметно плавное
уменьшение pH и заметен
скачок в области точки эквивалентности.
Скачок титрования полностью находится
в кислой области.
Точка эквивалентности расположена при
pH 5,28 и, очевидно, не
совпадает с точкой нейтральности. Скачок
титрования 0,1 М аммиака в пределах ±0,1%
от точки эквивалентности находится в
пределах pH от 6,25 до 4,30 и
составляет примерно 2 единицы pH,
что намного меньше скачка сильной щелочи
(6 единиц pH). С уменьшением
концентрации и увеличением температуры
скачок уменьшается.
В нашем случае одними из самых подходящих
являются лакмоид и хризоидин.
Окислительно-восстановительное титрование
Метод основан на реакциях
окисления-восстановления. Их называют
по применяемому тированному раствору
реагента, например: перманганатометрия,
йодометрия, бихроматометрия. В этих
методах в качестве титрантов применяют,
соответственно, KMnO4,
I2, K2Cr2O7.
В основе метода лежит изменение
окислительно-восстановительного
потенциала, обусловленного протеканием
окислительно-восстановительной реакции
между титрантом и определяемым веществом.
![]()
В процессе титрования происходит
изменение концентраций окисленной и
восстановленной форм, а, следовательно,
изменяется окислительно-восстановительный
потенциал титруемого раствора, включающей
две редоксопары.
В соответствии с уравнением Нернста
окислительно-восстановительный потенциал
для любой редоксопары:
![]()
Для каждого отдельного метода
окислительно-восстановительного
титрования используются свои стандартные
растворы.
Рассмотрим наш случай – перманганатометрия.
Рабочим раствором этого метода является
раствор перманганата калия KMnO4,
он неустойчив из-за реакции с водой,
катализируемый диоксидом марганца и
на свету:
![]()
Поэтому растворы перманганата калия
следует готовить, используя чистую воду
(органические примеси в воде могут
реагировать с
![]()
и давать MnO2, ускоряющий
разложение реагента), отфильтровать от
диоксида марганца и хранить в темных
склянках; раствор следует выдержать
несколько недель для окончания протекания
всех процессов. Очевидно, что раствор
следует стандартизировать, для чего
используют оксалат натрия и другие
восстановители. Реакция
![]()
катализируется ионами Mn2+.
Первые капли перманганата даже в горячем
растворе обесцвечиваются очень медленно.
В ходе титрования концентрация ионов
Mn2+ возрастает и
скорость реакции увеличивается: реакция
автокаталитическая.
Титр перманганата калия можно установить
также по оксиду мышьяка(III)
или металлическому железу.
В перманганатометрии применяют также
растворы восстановителей – слои Fe(II),
щавелевую кислоту и некоторые другие
– для определения окислителей методом
обратного титрования. Соединения Fe(II)
на воздухе медленно окисляются, особенно
в нейтральном растворе. Подкисление
замедляет процесс окисления, однако
обычно рекомендуется перед применением
раствора Fe(II)
в анализе проверить его титр. Оксалаты
и щавелевая кислота в растворе медленно
разлагаются. Этот процесс ускоряется
на свету, поэтому растворы оксалатов
рекомендуется хранить в темных склянках.
Подкисленные растворы оксалатов более
устойчивы, чем нейтральные или щелочные.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
1.
Окислительно-восстановительное
титрование
1
2.
Mэкв(X) = fэкв · М(X)
f = 1/z
z – число электронов, принимающих участие в
окислительно-восстановительной реакции
2
3.
10FeSO4 + 2KMnO4 + 8H2SO4 2MnSO4 +
5Fe2(SO4)3 + K2SO4 + 8H2O
MnO4 + 8H+ + 5ē Mn2+ + 4H2O
2Fe2+ 2ē Fe23+
3
4.
10FeSO4 + 2KMnO4 + 8H2SO4 2MnSO4 +
5Fe2(SO4)3 + K2SO4 + 8H2O
MnO4 + 8H+ + 5ē Mn2+ + 4H2O
2Fe2+ 2ē Fe23+
fэкв(KMnO4) = 1/5,
M(1/5KMnO4) = M(KMnO4) /5;
fэкв(FeSO4) = 1
M(FeSO4) = M(FeSO4)/1
4
5.
I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2ē 2I
2S2O32 2ē S4O62
5
6.
I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2ē 2I
2S2O32 2ē S4O62
fэкв(I2) = 1/2
M(1/2 I2) = M(I2)/2;
fэкв(Na2S2O3) = 1
M(Na2S2O3) = M(Na2S2O3)/1
6
7.
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
K2SO4 + 7H2O
Cr2O72 + 14H+ + 6ē Cr23+ + 7H2O
2I 2ē I2
7
8.
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
K2SO4 + 7H2O
Cr2O72 + 14H+ + 6ē Cr23+ + 7H2O
2I 2ē I2
fэкв(K2Cr2O7) = 1/6
Mэкв(K2Cr2O7) = M(K2Cr2O7)/6
fэкв(KI) = 1
M(KI) = M(KI)/1
8
9.
H2SO4
CH4
CH3—CH2OH
CH3OH
H
HCOH
H
H
CH3—COOH
H
H
H
9
10.
–4
–2
CH4
CH3OH
–3
–1
CH3—CH2OH
+1
HCOH
–3
-1
+3
CH3—COOH
10
11.
O O
OH OH
H
+1
H +2
+1
O
O
+2
CHOH
O
CHOH
CH2OH
CH2OH
C21+ – 2е
O
C22+
fэкв(Аск.к.) = 1/2
Mэкв(Аск.к.) = M(Аск.к.)/2
11
12.
Перманганатометрия
Титрант – 0,02 М или 0,1 М (1/5 KMnO4) или
0,1 н. раствор KMnO4
По точной навеске приготовить нельзя, т.к.
сильный окислитель.
Готовят раствор приблизительно нужной концентрации, выдерживают 7-10 дней или
кипятят 10 минут для окисления восстановителей, содержащихся в воде
Фильтруют через стеклянный фильтр.
12
13.
Стандартизацию проводят по щавелевой кислоте H2C2O4 2H2O, оксалату натрия Na2C2O4,
оксиду мышьяка (III) As2O3, металлическому
железу.
Стандартизацию проводят только в сернокислой
среде.
16HCl + 2KMnO4 5Cl2 + 2MnCl2 + 2KCl +8H2O
Индикатор – сам титрант.
13
14.
Стандартизация 0,1 н. по щавелевой кислоте
60-70 0С
5H2C2O4 + 2KMnO4 + 3H2SO4 2MnSO4 +
10CO2 + K2SO4 + 8H2O
C2O42– – 2е 2CO2
14
15.
Этикетка:
0,02 М или 0,1 М (1/5KMnO4) или 0,1 н.
К = 0,9972
Хранят в темном месте, в склянках темного
стекла
свет
4KMnO4 + 2H2O 4MnO2 + 4 KOH + 3H2O
По ГФ XIII стандартизация по тиосульфату
натрия
15
16.
Применение:
прямая перманганатометрия
Железа (II) сульфат:
10FeSO4 + 2KMnO4 + 8H2SO4 5Fe2(SO4)3 +
K2SO4 + 2MnSO4 + 8H2O
2Fe2+ – 2е Fe23+
Сэкв(KMnO4)·Mэкв(FeSO4)
Т(KMnO4/FeSO4) = ————————————
1000
16
17.
m(FeSO4) = V(KMnO4)·K·T(KMnO4/FeSO4)
V(KMnO4)·K·T(KMnO4/FeSO4)·100
(FeSO4) = ——————————————, %
а(FeSO4)
17
18.
Перекись водорода:
5H2O2 + 2KMnO4 + 3H2SO4 5O2 + K2SO4 +
2MnSO4 + 8H2O
O22– – 2е O2
Сэкв(KMnO4)·Mэкв(H2O2)
Т(KMnO4/H2O2) = ————————————
1000
18
19.
Аскорбиновая кислота:
O O
OH OH
H
H
5
+ 2KMnO4 + 3H2SO4
O
5
CHOH
O
CHOH
CH2OH
CH2OH
O
C21+ – 2е
+ 2MnSO4 + K2SO4 + 8H2O
O
C22+
Сэкв(KMnO4)·Mэкв(Аск.к.)
Т(KMnO4/Аск.к.) = ————————————
1000
19
20.
обратная перманганатометрия
Натрия нитрит:
5NaNO2 + 2KMnO4(изб.) + 3H2SO4 5NaNO3 +
2MnSO4 + K2SO4 + 3H2O
2KMnO4(ост.) + 10KI + 8H2SO4 2MnSO4 + 5I2
+ 6K2SO4 + 8H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
20
21.
(VKMnO4 K VNa2S2O3 K) Т(Na2S2O3/NaNO2) 100
(NaNO2)= (%)
а (NaNO2)
Сэкв(Na2S2O3) Мэкв(NaNO2)
Т(Na2S2O3/NaNO2) =
1000
NO2– + H2O – 2е
N3+ – 2е
NO3– + 2H+
N5+
21
22.
5NaNO2 + 2KMnO4(изб.) + 3H2SO4 5NaNO3 +
2MnSO4 + K2SO4 + 3H2O
NO2– + H2O – 2е NO3– + 2H+
| 5
MnO4– + 8H+ + 5е Mn2+ + 4H2O | 2
5NO2– + 2MnO4– + 16H+ + 5H2O
5NO3– + 10H+ + 2Mn2+ + 8H2O
или
5NO2– + 2MnO4– + 6H+ 5NO3– + 2Mn2+ + 3H2O
22
23.
Кривые окислительно-восстановительного
титрования
Выражают зависимость величины потенциала
от концентрации титранта
23
24.
Задача. Рассчитать и построить кривую титрования соли Fe (II) раствором перманганата
калия, если
Eo (Fe3+/Fe2+) = + 0,77 B;
Eo (MnO4–/Mn2+) = + 1,51 B
Какой из индикаторов можно использовать в
данном случае?
24
25.
2,2 – Дипиридил (комплекс с рутением)
Ео = 1,33 В
Дифениламин-2,2 –дикарбоновая кислота
Ео = 1,26 В
Комплекс 1,10-фенантролина с Fe (II)
Ео = 1,14 В
Фенилантраниловая кислота Ео = 1,08 В
Дифениламин
Ео = 0,76 В
Метиленовый голубой
Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
25
26.
5Fe2+ + MnO4– + 8H+ 5Fe3+ + Mn2+ + 4H2O
В начале титрования и до т.э. в растворе будут:
Fe2+, Fe3+ ,Mn2+
Потенциал системы будет определяться парой
восстановителя Fe3+/Fe2+
Величина потенциала может быть вычислена по
уравнения Нернста:
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
26
27.
В точке эквивалентности:
E = (mEoв-ля + nEoок-ля )/(m + n)
27
28.
5Fe2+ + MnO4– + 8H+ 5Fe3+ + Mn2+ + 4H2O
за т. э. в растворе будут:
Fe3+, MnO4– и Mn2+
Потенциал будет определяться парой
окислителя MnO4– /Mn2+
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/5 lg[MnO4–][H+]8/[Mn2+]
28
29.
Пусть концентрация ионов водорода равна 1 М
Доб-но
От-но
KMnO4–
в%
[Fe3+]
———
[Fe2+]
9
9
9/91 10-1
-1
0,77–0,059=0,71В
50
50
50/50=1
0
0,77 В
99
99
99/1 102
2
0,77+0,059 2=0,89В
3
0,77+0,059 3=0,95В
99,9
100
99,9 99,9/0,1 103
[Fe3+]
lg ———
[Fe2+]
E=Eo +0,059/1
lg [Fe3+]/[Fe2+]
E = (mEoв-ля+nEoок-ля )/(m+n)=(1 0,77+5 1,51)/(1+5)
=1,39 29B
30.
Доб-но
Изб-к
KMnO4–
в%
100,1
0,1
[MnO4–][H+]8
————— lgдроби E=Eo +0,059/5lgдр
[Mn2+]
0,1/100 10-3 -3
1,51+(0,059/5) (-3) =
1,475 B
101
1
1/100=10-2
-2
1,51+(0,059/5) (-2)
= 1,486 B
110
10
10/100=10-1
-1
1,51+(0,059/5) (-1)
= 1,498 B
30
31.
Скачок от 0,95 В до 1,475 В
2,2 – Дипиридил (комплекс с рутением)
Ео = 1,33 В
Дифениламин-2,2 –дикарбоновая кислота
Ео = 1,26 В
Комплекс 1,10-фенантролина с Fe (II)
Ео = 1,14 В
Фенилантраниловая кислота Ео = 1,08 В
Дифениламин Ео = 0,76 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
31
32.
Интервал изменения окраски индикаторов
Ind(ок) + ne Ind(вос)
0,059
[Ind(ок)]
E = Eo + ——— lg ————
n
[Ind(вос)]
[Ind(ок)]
———— = 10 – наблюдаем окраску окисленной
[Ind(вос)]
формы
E = Eo + 0,059/n lg10 = Eo + 0,059/n
32
33.
[Ind(ок)]
——— = 0,1 – наблюдаем окраску восстановл.
[Ind(вос)]
формы
E = Eo + 0,059/n lg 0,1 = Eo – 0,059/n
E = Eo ± 0,059/n
33
34.
Индикаторы окислительно-восстановительного
титрования
1. Специфические – индикаторы, которые взаимодействуют с одной из форм окислительновосстановительной пары с изменением
окраски (крахмал)
2. Редокс-индикаторы — вещества, которые при
определенном потенциале раствора окисляются или восстанавливаются с изменением
окраски
34
35.
Редокс-индикаторы:
1. Обратимые
E = Eo ± 0,059/n
Дифениламин Ео = 0,76 В
бесцветный – фиолетовый
м.р. в воде, готовят 1% р-р в концентр. H2SO4
Ферроин – комплекс Fe(II) с о-фенантролином
Ео = 1,14 В
[FeL3]2+ – e [FeL3]3+
красная
бледно-голубая
Фенилантраниловая кислота (Ео = 1,08 В) и др.
35
36.
2. Необратимые
Метиловый оранжевый
Метиловый красный,
Нейтральный красный
При окислении необратимо исчезает окраска
раствора
36
37.
Индикаторные ошибки окислительновосстановительного титрования
Индикаторная ошибка рассчитывается по
формуле:
n’(X)
Х = ———— · 100
n(X)
n’(X) – количество неоттитрованного вещества
(или избыточно прибавленного титранта)
n(X) – количество вещества, взятого для
титрования
37
38.
Задача. Рассчитать ошибку титрования железа
сульфата (II) раствором KMnO4 в сернокислой
среде при [H+] = 1 моль/л с индикатором
дифениламином (Ео = 0,76 В).
Потенциал системы в т.э.:
E = (mEoв-ля+nEoок-ля )/(m+n)=(1 0,77+5 1,51)/(1+5)
=1,39 B
38
39.
Таким образом,
Изменение окраски произойдет при 0,76 В
Т.э. наступит при 1,39 В, следовательно,
раствор недотитрован.
Потенциал системы рассчитывался по паре
[Fe3+]/[Fe2+] (Eo = 0,77 В) и закончили
титровать при 0,76 В
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
0,76 = 0,77 + 0,059/1 lg[Fe3+]/[Fe2+]
39
40.
lg[Fe3+]/[Fe2+] = – 0,169
[Fe3+]
0,68
———— = 0,68 = ———
[Fe2+]
1
n’(X)
Х = ———— · 100
n(X)
n’(X) = 1 (количество неоттитрованного в-ва)
n(X) = 1 + 0,68 (количество вещества, взятого
для титрования)
40
41.
Ошибка титрования составит:
1
Х = ———— · 100 = 59,5 %
1 + 0,68
Индикаторная ошибка д.б. не более 0,1 %.
41
42.
Задача. Рассчитать и построить кривую
титрования соли олова (II) раствором соли
железа (III), если Eo (Fe3+/Fe2+) = + 0,77 B и
Eo (Sn4+/Sn2+) = + 0,15 B.
Какой из индикаторов вы выберете?
Дифениламин Ео = 0,76 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
Нейтральный красный Ео = 0,24 В
Рассчитать ошибку титрования с индикатором –
метиленовый голубой
42
43.
Sn2+ + 2Fe3+ Sn4+ + 2Fe2+
В начале титрования и до т.э. в растворе будут:
Sn2+, Sn4+, Fe2+
Потенциал системы будет определяться парой
восстановителя Sn4+/Sn2+,
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/2 lg[Sn4+]/[Sn2+]
43
44.
В точке эквивалентности:
E = (mEoв-ля + nEoок-ля )/(m + n)
E = (2 · 0,15 + 1· 0,77) / 3 = 0,36 B
44
45.
Sn2+ + 2Fe3+ Sn4+ + 2Fe2+
за т. э. в растворе будут:
Fe3+, Sn4+, Fe2+
Потенциал будет определяться парой
окислителя Fe3+/Fe2+
Величина потенциала вычисляется по
уравнению Нернста:
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
45
46.
Доб-но
Fe3+
От-но
в%
[Sn4+]
———
[Sn2+]
[Sn4+]
lg ———
[Sn2+]
E
9
9
9/91 10-1
-1
0,12 В
50
50
50/50 1
0
0,15 В
90
90
90/10 101
1
0,18 В
99
99
99/1 102
2
0,21 В
99,9
99,9
99,9/0,1 103
3
0,24 В
100
0,36 B
46
47.
Доб-но
Fe3+
Изб-к
в%
100,1
0,1
0,1/100 10-3
-3
0,59 B
101
1
1/100=10-2
-2
0,65 B
110
10
10/100=10-1
-1
0,71 B
[Fe3+]
———
[Fe2+]
[Fe3+]
lg ———
[Fe2+]
E
Скачок от 0,24 В до 0,59 В
Метиленовый голубой Ео = 0,53 В
Индиготетрасульфоновая кислота Ео = 0,36 В
Нейтральный красный Ео = 0,24 В
47
48.
Изменение окраски произойдет при 0,53 В
Т.э. наступит при 0,36 В, следовательно,
раствор перетитрован.
Потенциал системы рассчитывался по паре
окислителя [Fe3+]/[Fe2+] (Eo = 0,77 В)
Закончили титровать при 0,53 В
E = Eo + 0,059/1 lg[Fe3+]/[Fe2+]
0,53 = 0,77 + 0,059/1 lg[Fe3+]/[Fe2+]
48
49.
lg[Fe3+]/[Fe2+] = – 6,949
[Fe3+]
1·10–7
———— = 1·10–7 = ———
[Fe2+]
1
1·10–7
Х = ——— · 100 = 1·10–5 %
1
n’(X) = 1·10–7 (количество избыточно прибавленного титранта)
n(X) = 1 + 1·10–7 1 (количество вещества,
взятого для титрования)
49
50.
Дихроматометрия
Титрант – 0,0167 М или 0,1 М (1/6 K2Cr2O7)
или 0,1 н. раствор K2Cr2O7
Стандартный раствор можно приготовить по
точной навеске
Cr2O72 + 14H+ + 6ē 2Cr3+ + 7H2O
fэкв(K2Cr2O7) = 1/6
Mэкв(K2Cr2O7) = M(K2Cr2O7)/6
50
51.
K2Cr2O7 – оранжевая окраска
Cr3+ – зеленоватая окраска, однако
интенсивности цвета не хватает для фиксации
конечной точки титрования
Ind – дифениламин, дифениламинсульфоновая
кислота и др.
51
52.
а(K2Cr2O7)теор=Сэкв(K2Cr2O7)·Mэкв(K2Cr2O7)·V (л)
а(K2Cr2O7)прак
Сэкв(K2Cr2O7)прак = —————————
Mэкв(K2Cr2O7) · V(л)
Сэкв(K2Cr2O7)прак
K = ————————
Сэкв(K2Cr2O7)теор
52
53.
Применение:
прямая дихроматометрия
Железа (II) сульфат:
6FeSO4 + K2Cr2O7 + 7H2SO4 3Fe2(SO4)3 +
Cr2(SO4)3 + K2SO4 + 7H2O
2Fe2+ – 2е Fe23+
Сэкв(K2Cr2O7)·Mэкв(FeSO4)
Т(K2Cr2O7/FeSO4) = ———————————
1000
53
54.
Расчет массы и массовой доли (%)
m(FeSO4) = V(K2Cr2O7)·K·T(K2Cr2O7/FeSO4)
V(K2Cr2O7)·K·T(K2Cr2O7/FeSO4)·100
(FeSO4) = ——————————————, %
а(FeSO4)
54
55.
Калия иодид:
6KI + K2Cr2O7 + 7H2SO4 3I2 + Cr2(SO4)3 +
4K2SO4 + 7H2O
2I– – 2е I2
Сэкв(K2Cr2O7)·Mэкв(KI)
Т(K2Cr2O7/KI) = ——————————
1000
55
56.
Кислота аскорбиновая:
O O
OH OH
H
3
O
+ K2CrO4 + 4H SO
2
4
O
H
CHOH
O
CHOH
CH2OH
CH2OH
C21+ – 2е
+ Cr2(SO4)3 + K2SO4 + 7H2O
O
C22+
С(1/6K2Cr2O7)·M(1/2Аск.к.)
Т(K2Cr2O7/Аск.к.) = ———————————
1000
56
57.
Обратная дихроматометрия применяется для
определения спирта этилового:
3CH3CH2OH + 2K2Cr2O7 + 8H2SO4
избыток
3CH3COOH + 2Cr2(SO4)3 + 2K2SO4 + 11H2O
K2Cr2O7 + 6KI + 7H2SO4 3I2 + Cr2(SO4)3 +
остаток
4K2SO4 + 7H2O
I2 + 2Na2S2O3 2NaI + Na2S4O6
57
58.
Расчет массовой доли (%)
(V(K2Cr2O7) K V(Na2S2O3) K) Т(Na2S2O3/Сп) 100
(Сп) = (%)
а (CH3CH2OH)
Сэкв(Na2S2O3) Мэкв(Спитра)
Т(Na2S2O3/Сп) =
1000
-1
CH3—CH2OH – 4 е
+3
CH3—COOH
58
59.
Преимущества дихроматометрии перед
перманганатометрией
1. Титрант можно приготовить по точной
навеске
2. Титрование можно проводить в присутствии
HCl
Eo(Cr2O72–/2Cr3+) = 1,33 B
Eo(Cl2/2Cl–) = 1,33 B
При титровании Cl– не окисляются Cr2O72–ионами
59
Чтобы окраска окислительно-восстановительного индикатора изменялась при титровании резко и индикаторная ошибка титрования была незначительной, интервал перехода индикатора должен находиться в пределах скачка потенциалов на кривой титрования. [c.369]
И. М. Кольтгоф, В. А. Стенгер. Объемный анализ. Госхимиздат, 1950, (т. I. 376 стр.) и 1952, (т. И, 444 стр.). В т. I рассматриваются теоретические основы объемного анализа. Изложена теория методов нейтрализации и соединения ионов, приведены кривые титрования для различных случаев метода нейтрализации. Отдельные главы содержат материал ио теории методов окисления-восстановления, теории индикаторов, по ошибкам титрования. Рассмотрены явления адсорбции и соосаждения, катализа и индукции, применение объемных методов в органическом анализе описаны теоретические положения, касающиеся применения физико-химических методов для определения точки эквивалентности. В т. 11 книги изложено практическое применение методов нейтрализации, осаждения и комплексообразования. В томе 111 (840 стр., 1961 г.) описано применение окислительно-восстановительных методов объемного анализа. [c.486]
Инцикаторы в окислительно-восстановительном титровании. Индикаторные ошибки титрования [c.132]
Ошибка титрования близка нулю, если потенциал перехода окраски индикатора равен потенциалу в точке эквивалентности. Практически допустимой считают относительную ошибку в10 или 0,1 7о, благодаря чему возможен широкий выбор окислительно-восстановительных индикаторов. При титровании восстановителя 2 окислителем 1 предельные значения потенциа-.лов (в вольтах) получают из следующих уравнений [c.170]
Кривые титрования хлоридов нитратом серебра в расплавленных солях получаются такими же, как кривые титрования в водных растворах для экзотермических процессов. Относительная ошибка титрования для указанных выше концентраций приближалась к 4%. Применение метода ограничено в связи с трудностью достаточно точного измерения повышения температуры, происходящего в результате реакции. Необходимо, чтобы изменение температуры окружающей среды было на несколько порядков меньше, чем изменение температуры, происходящее в результате рассматриваемой реакции. Теоретически в среде расплавленных солей возможно проводить любые реакции осаждения, комплексообразования или окислительно-восстановительные реакции, однако практически сложность прибора, который требуется для этого, ограничивает применимость расплавленных солей в качестве растворителей. [c.110]
На практике расчет потенциала при помощи такого уравнения, как правило, связан с ошибкой, хотя во многих случаях эта ошибка невелика и полученное значение потенциала является полезным для предсказания возможности проведения того или иного титрования. Во всех окислительно-восстановительных системах, включающих ионы водорода, потенциал обычно зависит от концентрации этих ионов, однако степень такой зависимости может значительно отличаться от величины, полученной по уравнению. Такая же картина иногда наблюдается и для зависимости потенциала от концентрации окисленной и (или) восстановленной формы вещества. [c.353]
Разница в объемах постоянна для всех выбранных на кривой точек, до которых проводилось титрование, и является, вероятно, приборной ошибкой. Результаты титрования до заданного потенциала показывают, что при помощи автотитратора можно приготавливать растворы с заранее заданными величинами окислительно-восстановительного потенциала или pH, что позволяет автоматизировать процесс подготовки проб к дальнейшему анализу при серийных определениях. [c.136]
Для окислительно-восстановительного титрования перманганата применяют также другие восстановители, в том числе соединения Си, Sn , W», U , V [17], однако преимущества применения этих восстановителей не очевидны. Сравнительно простой окислительно-восстановительный метод основан на взаимодействии перманганата с кислым раствором KI и последующем титровании выделившегося иода стандартным раствором тиосульфата. Избирательность определения часто достигается применением маскировки. Например, в указанном выше методе перманганат (или хромат) можно определять в присутствии железа (III), которое маскируют гексаметафосфатом [18]. При определении 13—65 мг марганца (в виде MnO ) в присутствии 10—340 мг Fe + ошибка составляет 0,3%. [c.159]
В Присутствии восстановителей-абсорбентов в ячейке для поглощения. Осуществить такое определение с хлор- или бромсеребряными электродами не представляется возможным, так как они дают ошибку, связанную с возникновением на них окислительно-восстановительного потенциала. Сжигание проводится обычным способом, потенциометрическое титрование идет в 50%-ном диоксане. Титруют 0,02 М раствором нитрата серебра. Вблизи конечной точки титрант вводят равными порциями по 0,02 мл. Конечную точку определяют как максимальное значение Д /Д . [c.62]
Амперометрическое титрование применимо для многих окислительно-восстановительных реакций, реакций комплексообразования и осаждения. По сути этот метод обладает более высокой точностью, чем соответствующий прямой полярографический метод, так как каждое определение включает ряд отдельных измерений, для которых исключаются случайные ошибки. [c.359]
Преимущество флуоресцентных окислительно-восстановительных индикаторов по сравнению с цветными состоит в том, что с их применением можно значительно уменьшить ошибки титрования, связанные с расходом титранта на окисление индикатора. Кроме того, возможно титрование в окрашенных и мутных средах . [c.120]
Окислительно-восстановительные реакции в большинстве случаев протекают в сильно кислой среде небольшие изменения электропроводности за счет реакции окисления-восстановления на фоне высокой проводимости среды обусловливают нерезкие перегибы кривой титрования в конечной точке. Это приводит к значительным ошибкам в определении конечной точки. В работе [69] систематизированы реакции этого типа в наглядной таблице для целей кондуктометрического титрования. Эта таблица может быть использована при разработке методов ВЧ-титрования окислителей и восстановителей. [c.156]
Последние соотнощения показывают, что в состоянии равновесия концентрация Мп2+ будет превыщать концентрацию МпО в 1025 раз . р g процесс восстановления ионов МпО в Мп2+ пройдет с исчерпывающей для практической цели полнотой. Поэтому можно считать, что ощибка титрования, обусловленная образованием равновесной системы, будет равна нулю (такое количественное течение процесса вообще характерно для большинства окислительно-восстановительных реакций). Ввиду этого ошибки, допускаемые при перманганатометрических определениях, должны быть отнесены лишь за счет капельной ошибки, неточностей при отсчетах и взвешиваниях, а также необходимости введения некоторого избытка перманганата, позволяющего установить конец реакции по появлению окраски раствора. [c.85]
Практикум содержит 47 лабораторных заданий. Многие задания оригинальны по постановке и рассчитаны на получение количественных результатов. Некоторые из задач могут быть использованы как факультативные. Новые работы посвящены методам очистки веществ ионитами и зонной плавкой, определению термодинамических характеристик процесса растворения бензойной кислоты и процесса восстановления ионов меди цинком, определению координационного числа методами криоскопии и титрования, кинетике окислительно-восстановительных реакций и изучению поверхностно-активных веществ. Заново написано задание по определению тепловых эффектов. В новом издании практикума лабораторным заданиям предпослано введение, где рассказывается о научном эксперименте и его роли в познании, и глава о работе с экспериментальными данными, в которой идет речь о записи результатов, вычислениях, ошибках эксперимента, выражении результатов в виде графиков и формул и о написании отчета. Остальные задания практикума подверглись значительной переработке. В приложении появился ряд новых таблиц. [c.3]
В книге на современном уровне кратко изложены теоретические основы гравиметрии и титриметрии — образование и свойства осадков, типы химических равновесий в гомогенных и гетерогенных растворах описаны кривые титрования проанализированы ошибки в кислотно-основном, осацительном, комплексимет-рическом и окислительно-восстановительном титровании. Подробно рассмотрены аппаратура и техника проведения всех операций в количественном химическом анализе. Все расчеты проведены с учетом новых данных о величинах констант, стандартных потенциалов и т.п. [c.2]
Окислительно-восстановительный потенциал аскорбиновой кислоты при pH 7 составляет — -0,19 в. Аскорбинометрическое определение железа является одним из лучших методов, так как не мешают нитраты и фосфаты присутствие фторидов вызывает незначительную ошибку. Недостатком является малая устойчивость титрованного раствора аскорбиновой кислоты при хранении. [c.193]
Кинетическое окислительно-восстановительное титрование Sb(lII). Реакционную смесь титруют раствором окислителя (КВгОз, Ja, e(S04)2, K rjO,, KJO3) в строго определенных условиях (pH, температура, объем) при постоянной скорости подачи титранта с потенциометрическим, фотометрическим или визуальным (индикаторы ксиленоловый оранжевый, ферроин) установлением конечной точки. По продолжительности титрования, которое прямо пропорционально содержанию Sb, находят ее содержание. Метод позволяет определять Sb в растворах с ее концентрацией 8-10 —1,2-10 с ошибкой 2—5% [953, 1326]. [c.98]
Эрдеи и Ради [938] титровали Au(III) аскорбиновой кислотой при pH 1—3 и 50—60° С. При температуре > 80° С получаются заниженные результаты. Не мешают Hg(II), u, Fe(III) (в присутствии НэР04), 150-кратные количества NOJ, 1 г-ион л С мешают ионы со стандартным окислительно-восстановительным потенциалом > — — 1,39 в [Pt(IV), Вг , S N и N ]. Ошибка определения золота в 0,001—0,01 N растворах 1%. При титровании в среде ледяной уксусной кислоты [937] вид кривой титрования похож на кривую титрования в водных растворах, если перед титрованием в безводной уксусной кислоте ввести безводный Ha OONa. В точке эквивалентности наблюдается отчетливый скачок потенциала. Аналогично золоту ведут себя другие окислители. [c.130]
ВОЗМОЖНОЙ благодаря снижению окислительно-восстановительного потенциала пары Fe +/Fe + за счет образования устойчивых пирофосфатных комплексов Ее(П1) при pH 9, а в начале титрования — при pH 12 [915]. Титрование ведут 0,01 JV раствором ЕеЗОд в инертной атмосфере с обязательным добавлением в анализируемый раствор бромид-иона, препятствующего диспропорцио-нированию гипобромит-иона. 1 —10 мг ВгО определяют этим методом с ошибкой -< 0,5%. Примеси BrOJ (до 16%), 10-кратное количество ионов СГ или J , 10-кратный избыток сульфат-иона и 150-кратный нитрат-иона не мешают анализу. [c.129]
При прямом титровании фосфатов раствором соли свинца [1172] при рн 2—3 в качестве индикатора применяют хлороформный раствор дитизона. Титруют до перехода зеленой окраски в фиолетовую. Метод применяют для определения фосфора в фосфатных удобрениях [1174]. В качестве индикатора применяют также эриох-ром черный Т (растворяют 0,2 г эриохрома черного Т в 5 мл С2Н5ОН и 15 мл триэтаНоламина) [950]. Титруют до появления красной окраски. Метод применяют для определения микроколичеств фосфора в органических веществах. Для определения микроколичеств фосфора применяют также титрование нитратом свинца в присутствии 2-азо-4-резорцина [1018]. Титруют до появления красного окрашивания. При содержании фосфора 20— 400 мкг средняя абсолютная ошибка определения составляет 2—3 мкг Р. При косвенном определении фосфатов с помощью нитрата свинца применяют окислительно-восстановительные индикаторы [732, 733]. Метод основан на осаждении РО/ в виде РЬз(Р04)2 нитратом свинца, избыток которого оттитровывают К4[Ге(СК)б1 в присутствии вариаминового синего и Кз[Ге(СК)б] в качестве индикатора. Титруют до перехода фиолетовой окраски в бледно-желтую. [c.37]
Предложен интересный метод окислительно-восстановительного титрования, основанный на применении гидрохинона [20]. Метод позволяет определять 10—130 мг перманганата в присутствии других окислителей, например, бихромата, гексацианоферрата (III) и хлорамина Т. При определении 13—130 мг КМПО4 в присутствии 20—2000 мг других окислителей относительная ошибка оп-)еделения не превышает 1,8%. Определению мешает ванадий (V). Метод длителен сначала перманганат восстанавливают до диоксида марганца с помощью формиата натрия в щелочном растворе. Осадок гидратированного диоксида марганца фильтруют, промывают, растворяют в ЫагНгРгОу и образующийся пирофосфат марганца (III) титруют стандартным раствором гидрохинона. [c.159]
Хотя растворы бихромата окрашены в оранжевый цвет, интенсивность окраски недостаточна для определения конечной точки. Отличным индикатором при титровании бихроматом является дифениламиносульфокислота (гл. 15) с переходом окраски от зеленой (ионы хрома(1П)) до фиолетовой (окисленная форма индикатора). Холостой опыт в присутствии индикатора идет плохо, поскольку в отсутствие других окислительно-восстановительных систем бихромат очень медленно окисляет индикатор. Но ошибка, возникающая за счет того, что холостой опыт не проведен, обычно пренебрежимо мала. Реакция окисления дифениламиносульфокис-лоты обратима, поэтому можно проводить обратное титрование очень малых количеств бихромата железом (II). В присутствии больших количеств окислителя при низкой кислотности (рН>2) раствора индикатор необратимо окисляется до соединений желтого или красного цвета. [c.386]
Метод определения конечной точки. Как уже отмечалось, перманганат-ион имеет настолько интенсивный цвет, что сам по себе может служить индикатором. Опыты показали, что при титровании мышьяковистой кислоты перманганатом концентрация иона МпОГ. равная б-10 моль/л, легко обнаруживается вблизи точки эквивалентности. В расчетах, проведенных для одного из опытов титрования, мы показали, что при введении 0,1% избытка титранта концентрация МпОГ составляет 8-10 моль/л. Таким образом, ясно, что при использовании цвета избыточного Мп04 для установления точки эквивалентности вводится ошибка менее 0,1 %. Однако можно подобрать окислительно-восстановительный индикатор, при применении которого конечная точка еще точнее соответствовала бы точке эквивалентности. В рассматриваемом случае важнее даже то, что при использовании индикатора конечную точку можно обнаружить еще в тот момент, когда концентрация МпОГ слишком низка, чтобы окислить хлорид-ионы, присутствующие в растворе. Один из таких индикаторов — о-фенаитролиповый комплекс желе-за П), с которым могут происходить следующие превращения [c.235]
Работа установки проверялась не только титрованием в кислотно основных и окислительно-восстановительных сис темах, но также пу тем определения миллиграммовых количеств галогенид ионов их осаждением ионами серебра, г енерированнымн электрохимически. Определения проводят в бюксе на 75 мл, анодом служит серебро вы сокой чистоты или платина, покрытая серебром, катодом — платиновая спираль, снабженная чехлом. В качестве индикаторного электрода используют серебряную или платиновую проволоку, потенциал ко торой измеряется относительно каломельного электрода сравнения, причем для устранения загрязнения хлоридом последний под.-оединя-н.т с помощью агарового солевого мостика- с 0,1 М раствором а. 0,. При определении хлорида, бромида и иодида в количествах 0,2 — 10 средняя ошибка составляет 0,005 мг. [c.86]
Окислительно-восстановительное титрование
— использует реакции, протекающие с изменением степени окисления реагирующих веществ.
Каждую окислительно-восстановительную реакцию можно представить как сумму двух полуреакций. Одна реакция отражает превращение окислителя, а вторая – восстановителя:
Ox1+ ne— = Red1
Red2 – ne— = Ox2
Молярная масса эквивалента окислителя или восстановителя в реакции зависит от числа принятых или отданных одной молекулой окислителя (восстановителя) электронов. Молярная масса эквивалента окислителя (восстановителя) равна произведению фактора эквивалентности данного вещества на его молярную массу:
M (fэкв (В)В) = fэкв (В) М (В),
где M (fэкв (В)В) – молярная масса эквивалента вещества В
М (В) – молярная масса вещества В
fэкв (В) – фактор эквивалентности вещества.
fэкв (В) = 1/n,
n – число электронов, принятых или отданных одной молекулой окислителя (восстановителя) в данной реакции.
Требования к реакциям в Red-Ox-метрии
К реакциям, применяемым в окислительно-восстановительном титровании, предъявляются следующие требования:
а) Реакции должны протекать до конца, являться необратимыми, не сопровождаться побочными процессами.
б) В ходе реакции должны образовываться продукты определенного известного состава.
в) Реакции должны протекать быстро.
г) Должна быть возможность фиксировать точку эквивалентности.
К недостатком окислительно-восстановительных реакций в большинстве случаев относится их невысокая скорость, что затрудняет процесс титрования. Для ускорения реакций применяют нагревание. Если нагревание использовать нельзя (вещество разлагается или улетучивается), то увеличивают концентрацию вещества или используют катализаторы.
Окислительно-восстановительный потенциал и факторы, влияющие на него
Известно, что химическая реакция протекает только в том случае, если изменение энергии Гиббса ∆Gr < 0. Для любой окислительно-восстановительной реакции справедливо термодинамическое соотношение:
∆Gr = — n · F · ЭДС,
где n — число отданных или присоединенных электронов;
F — постоянная Фарадея (96500 Кл/моль);
ЭДС = Eокисл. – Е восст. — разница в значениях окислительно-восстановительных потенциалов окислителя и восстановителя.
Тогда необходимым условием протекания реакции является ЭДС > 0.
Окислительно-восстановительный потенциал или редокс-потенциал – это количественная характеристика окислительно-восстановительной реакции.
Окислительно-восстановительный потенциал E или φ зависит от природы окислительно-восстановительной пары, концентрации (активности) ионов окислителя и восстановителя, температуры. Количественно зависимость от этих параметров определяется уравнением Нернста:
ЕOx/Red = Е0Ox/Red + (0,059/n) lg([Ox]a / [Red]b),
где ЕOx/Red — окислительно-восстановительный потенциал пары Ox — Red, В;
Е0Ox/Red — стандартный окислительно-восстановительный потенциал пары Ox — Red, В;
([Ox] и [Red] — молярная концентрация окисленной и восстановленной форм соответственно, моль/л,
a и b — стехиометрические коэффициенты.
Если реакция протекает с участием молекул или ионов среды, то их концентрации также вводят в уравнение Нернста. Так для полуреакции MnO4— + 8H+ +5e— → Mn2+ + 4H2O уравнение Нернста может быть записано следующим образом:
Е MnO4-/ Mn2+ = Е0 MnO4-/ Mn2+ + (0,059/5) lg([MnO4—] · [H+]8 / [Mn2+]).
Реальный потенциал редокс-пары титрантов окислителей должен иметь значение потенциала на 0,4 – 0,5 В выше, чем потенциал редокс-пары титруемого восстановителя [(Eокисл. – Е восст) > 0,4], только в таком случае выполняются требования к реакциям в Red-Ox-метрии. Для регулирования потенциала редокс-пар титранта и определяемого вещества используют изменение рН среды, комплексообразующие добавки, увеличение температуры и т.д.
Равновесный окислительно-восстановительный потенциал зависит от ряда факторов:
1) От рН среды. Стандартный окислительно-восстановительный потенциал для приведенной выше реакции Е0 MnO4-/ Mn2+ = 1,51 B. С увеличением рН раствора окислительно-восстановительный потенциал этой пары будет уменьшаться.
2) От концентрации (активности) окисленной и восстановленной форм окислителя или восстановителя. С изменением концентраций (активностей) окисленной и восстановленной форм величина редокс-потенциала может изменяться. Например, для пары Fe3+/Fe2+ при условии [Fe3+] = [Fe2+] стандартный окислительно-восстановительный потенциал равен 0,77 В. Уравнение Нернста для полуреакции Fe3++ 1e → Fe2+ имеет вид:
Е Fe3+/Fe2+ = Е0 Fe3+/Fe2+ + 0,059 lg ([Fe3+] / [Fe2+]).
Изменяя концентрации окисленной или восстановленной форм вещества можно изменить величину редокс-потенциала.
Пример 1. Если в раствор, содержащий ионы Fe3+ и Fe2+ добавить SnCl2, проявляющий восстановительные свойства, то в растворе в результате реакции
2Fe3+ + Sn2+ →2Fe2+ + Sn4+ уменьшится концентрация ионов Fe3+ и увеличиться концентрация ионов Fe2+. При этом редокс-потенциал пары Fe3+/Fe2+ уменьшится Е Fe3+/Fe2+ = Е0 Fe3+/Fe2+.
Пример 2. При добавлении в раствор, содержащий ионы Fe3+ и Fe2+ раствора MnO4—, имеющего свойства окислителя, в результате реакции 5Fe2+ + 2MnO4— + 8H+ → 5Fe3++ 2Mn2+ + 4H2O
произойдет уменьшение концентрации ионов Fe2+ и увеличение концентрации ионов Fe3+. Следовательно, величина равновесного окислительно-восстановительного потенциала увеличиться Е Fe3+/Fe2+ > Е0 Fe3+/Fe2+.
От процесса комплексообразования. Величина редокс-потенциала значительно изменяется, если окисленная или восстановленная форма вещества в анализируемом растворе участвует в процессе комплексообразования.
Потенциал редокс-пары, например, M3+/M2+ в отсутствии комплексообразования будет при 25 °С равен:
Е M3+/M2+ = Е0 M3+/M2+ — 0,059 lg([M3+] / [M2+]),
При комплексообразовании с лигандом Lz— концентрация ионов M3+ уменьшится:
M3+ + Lz— = ML3-z
Константа устойчивости ML3-z равна:
βML = [ML3-z] / ([M3+] · [Lz—]).
Из данного выражения концентрация ионов M3+
[M3+] = [ML3-z] / ([Lz—] · βML),
Подставив ее в исходное уравнение Нернста, после ряда преобразований получим:
Е M3+/M2+ = Е0 M3+/M2+ — 0,059 lg βML
Пример 3. В присутствии фторид-ионов, образующих с ионами Fe3+ комплексный ион FeF2+ (lg β = 6,04) концентрация ионов Fe3+ резко падает и потенциал пары Fe3+ / Fe2+ уменьшается:
E Fe3+ / Fe2+ = 0,77 – 0,059 · 6,04 = 0,41 B.
За счет изменения потенциала можно изменить направление протекания реакции. Так при добавлении к раствору хлорида железа (III) раствора иодида калия между ними протекает реакция: 2FeCl3 + 2KI → 2FeCl2 + I2 + 2KCl
Стандартные окислительно-восстановительные потенциалы пар составляют:
E0 Fe3+ / Fe2+ = 0,77 B
E0 I2 / 2I— = 0,54 B
Тогда ЭДС = Eокисл. – Е восст = 0,77 – 0,54 = 0,23 В.
ЭДС > 0, реакция может протекать.
Если реакцию вести в присутствии фторид-ионов, то реакция протекать не будет, так как ЭДС < 0, (ЭДС = 0,41 – 0,54 = -0,13В)
FeCl3 + KI + F— ≠
4) От образования малорастворимых веществ. В присутствии ионов, способных образовывать малорастворимые соединения, потенциал окислительно-восстановительной пары можно вычислить следующим образом:
Е0 Me2+/MeX = Е Me2+/Me – 0,059/n lgПР(MeX) .
Пример 4. ЭДС реакции 2Cu2+ – 4I— → 2CuI + I2 без учета образования малорастворимого иодида меди (I) CuI равна
ЭДС = E0окисл. – Е0восст = E0 Cu2+ / Cu+ — E I2 / 2I— = 0,159 – 0,54 = -0,399B, ЭДС < 0
И, следовательно, реакция невозможна. Тогда как на самом деле данная реакция осуществима, т.к. с учетом образования малорастворимого иодида меди(I) CuI (lg ПР (CuI)=11,96) потенциал пары Cu2+ / CuI можно вычислить таким образом:
Е0 Cu2+ / CuI = 0,159 + 0,059 · 11,96 = 0,865 B
И ЭДС реакции 2Cu2+ – 4I— → 2CuI + I2
ЭДС = E0 Cu2+ / CuI — E0 I2 / 2I- = 0,865 – 0,54 = 0,325 B.
Таким образом, для данной реакции ЭДС > 0, поэтому реакция осуществима.
Классификация методов Red-Ox-метрии и способы титрования
Окислительно-восстановительное титрование или редоксиметрия (от латинского oxydatio — окисление и reductio — восстановление) основано на реакциях окисления-восстановления. Если титрант – окислитель, то титрование называют окислительным (оксидиметрия). Если титрант – восстановитель, то титрование восстановительное (редуциометрия)
По типу применяемого титранта методы окислительно-восстановительного титрования делятся на следующие виды:
• Пермангатометрическое – титрант раствор KMnO4;
• Иодометрическое титрование – титранты растворы I2 и Na2S2O3;
• Броматометрическое – титрант раствор KBrO3;
• Бромометрическое – титрант раствор Br2 (KBrO3 + KBr);
• Хроматометрическое – титрант раствор K2Cr2O7 и т.д.
Редокс-титрование может быть выполнено различными способами: прямое титрование, обратное титрование и заместительное титрование.
Прямое титрование проводят при ЭДС ≥ 0,4 В, что обеспечивает необходимую полноту и скорость протекания реакции.
Прямым титрованием можно определить:
а) ионы Fe2+ – титрант раствор KMnO4, ЭДС = 0,74 В:
10 FeSO4 + 2KMnO4 + 8H2SO4 = 5Fe2(SO4) 3 + 2MnSO4 + K2SO4 + 8H2O
В точке эквивалентности n (1/2 FeSO4) = n (1/2 KMnO4)
б) I2 – титрант раствор Na2S2O3 (ЭДС = 0,42 В):
I2 + 2Na2S2O3→ 2NaI + Na2S4O6
n (1/2 I2) = n (Na2S2O3)
Обратное титрование – используют при медленно протекающих реакциях. При этом к титруемой смеси добавляют избыток реагента (титранта 1) и выдерживают определенное время для полноты протекания реакции. Затем избыток реагента (титранта 1) оттитровывают другим титрантом 2.
Пример 5. При определении сульфидов добавляют раствор I2 (титрант 1), избыток которого затем оттитровывают раствором Na2S2O3 (титрант 2):
Na2S + I2 (изб.) + 2HCl = S + 2NaCl + 2HI
I2(ост) + 2Na2S2O3 = 2NaI + Na2S4O6
n(1/2 Na2S) = n (1/2 I2) — n (Na2S2O3)
Пример 6. Определение содержания свободного или связанного формальдегида в разнообразных технических продуктах:
HCHO + H2O2 → HCOOH + H2O
HCOOH + NaOH(изб) → HCOONa + H2O
NaOH(ост) + HCl → NaCl + H2O
N (1/2 HCHO) = n (NaOH) – n (HCl)
Заместительное титрование – определяют заместитель – продукт реакции, выделяющийся в эквивалентном количестве при взаимодействии определяемого вещества с каким-либо реагентом.
Таким образом, например, можно определять вещества, не вступающие в окислительно-восстановительные реакции.
Пример 7. Определение карбоната кальция:
CaCO3 + 2HCl → CaCl2 + CO2 + H2O
CaCl2 + (NH4) 2C2O4 → CaC2O4 + 2NH4Cl
CaC2O4 + H2SO4 → CaSO4 + H2C2O4
5H2C2O4 + 2KMnO4 + 2H2SO4 → 2MnSO4 + 10CO2 + 8H2O
В точке эквивалентности соблюдается равенство
N (1/2 CaCO3) = n (1/5 KMnO4)
Окислительно-восстановительные индикаторы
Для определения точки эквивалентности в Red-Ox-метрии используют различные индикаторы:
1) Окислительно-восстановительные индикаторы (редокс-индикаторы), изменяющие цвет при изменении окислительно-восстановительного потенциала системы.
2) Специфические индикаторы, изменяющие свой цвет при появлении избытка титранта или исчезновении определяемого вещества. Специфические индикаторы применяют в некоторых случаях. Так крахмал – индикатор на присутствие свободного йода, вернее трииодид-ионов I—3. В присутствии I—3 крахмал при комнатной температуре синеет. Появление синей окраски крахмала связано с адсорбцией I—3 на амилазе, входящей в состав крахмала.
Иногда в качестве индикатора используют тиоцианат аммония NH4SCN при титровании солей железа(III), катионы Fe3+с ионами SCN— образуют соединение красного цвета. В точке эквивалентности все ионы Fe3+ восстанавливаются до Fe2+ и титруемый раствор из красного становится бесцветным.
При титровании раствором перманганата калия сам титрант играет роль индикатора. При малейшем избытке KMnO4 раствор окрашивается в розовый цвет.
Редокс-индикаторы делятся на: обратимые и необратимые.
Обратимые индикаторы – обратимо изменяют свой цвет при изменении потенциала системы. Необратимые индикаторы – подвергаются необратимому окислению или восстановлению, в результате чего цвет индикатора изменяется необратимо.
Редокс-индикаторы существуют в двух формах окисленной (Indoкисл) и восстановленной (Indвосст.), причем цвет одной формы отличается от цвета другой.
Indoкисл + ne— ↔ Indвосст
Переход индикатора из одной формы в другую и изменение его окраски происходит при определенном потенциале системы (потенциале перехода). Потенциал индикатора определяется по уравнению Нернста:
EInd = E0Ind +0,059/n lg ([Indoкисл] / [Indвосст])
При равенстве концентраций окисленной и восстановленной форм индикатора EInd = E0Ind. При этом половина молекул индикатора существует в окисленной форме, половина – в восстановленной форме. Интервал перехода индикатора (ИП) лежит в пределах отношений концентраций обеих форм индикатора от 1/10 до 10/1.
При проведении окислительно-восстановительного титрования необходимо подбирать индикатор таким образом, чтобы потенциал индикатора находился в пределах скачка потенциала на кривой титрования. Многие индикаторы окислительно-восстановительного титрования обладают кислотными или основными свойствами и могут менять свое поведение в зависимости от рН среды.
Одним из наиболее известных и употребимых редокс-индикаторов является дифениламин (C6H5) 2NH.
Восстановленная форма индикатора бесцветная. Под действием окислителей дифениламин сначала необратимо переходит в бесцветный дифенилбензидин, который затем обратимо окисляется до сине-фиолетового дифенилбензидинфиолетового. EInd = 0,76 B.
Титрование индикаторным методом возможно, если для данной реакции ЭДС ≥ 0,4 В. При ЭДС = 0,4 – 0,2 В используют инструментальные индикаторы.
Кривые окислительно-восстановительного титрования
Кривую окислительно-восстановительного титрования представляют в виде зависимости потенциала E от количества добавленного титранта V (рис. 1 – А):
E = f (V)
Величина скачка в точке эквивалентности зависит от разности потенциалов двух окислительно-восстановительных пар, участвующих в процессе. Увеличение ЭДС приводит к возрастанию скачка титрования. Для увеличения ЭДС можно изменять концентрацию одного из компонентов редокспары. У окислителей можно повысить реальный потенциал редокс-пары, связав в комплекс восстановленную форму. Потенциал восстановителя можно понизить, связав в комплекс его окисленную форму.

Рис. 1. Кривые окислительно-восстановительного титрования:
А – интегральная кривая; В – дифференциальная кривая.
Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах ∆E/∆V—V (рис. 1 — Б). На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности. Определение точки эквивалентности по дифференциальной кривой значительно точнее, чем по простой зависимости E – V.
Перманганатометрия
В сильнокислой среде перманганат-ионы обладают высоким окислительно-восстановительным потенциалом, восстанавливаясь при этом до Mn(II). Поэтому перманганат калия применяют для определения многих восстановителей. Окисление восстановителей можно проводить в различных средах. Перманганат калия в кислой среде восстанавливается до ионов Mn2+, в нейтральной – до марганца(IV) или диоксида марганца MnO2, в щелочной среде – до марганца(VI) или манганата калия K2MnO4.
В методе перманганатометрии титрование чаще проводят в кислой среде:
MnO4— + 8H+ + 5e— → Mn2+ + 4H2O
E0 = 1,51 B; M(1/5 KMnO4) = 31,608 г/моль
Реже используют титрование в нейтральной среде:
MnO4— + 2H2O + 3e— → MnO2 + 4OH—
E0 = 0,60 B; M (1/3 KMnO4) = 52,68 г/моль
При титровании перманганатом не применяют индикаторы, так как титрант сам окрашен и является чувствительным индикатором: 0,1 мл 0,01 М раствора KMnO4 окрашивает 100 мл воды в бледно-розовый цвет.
Приготовление 0,1 н. (0,05 н.) раствора перманганата калия
Титрованный раствор перманганата калия по точной навеске кристаллического KMnO4 приготовить нельзя, так как в нем всегда содержится некоторое количество MnO2 и других продуктов разложения. Поэтому раствор перманганата калия относится к вторичным стандартным растворам. Первоначально готовят раствор KMnO4, концентрация которого приблизительно равна необходимой концентрации. Навеску берут на технохимических весах несколько больше расчетной величины. Так как KMnO4 является сильным окислителем и изменяет свою концентрацию в присутствии различных восстановителей, то приготовленный раствор перманганата калия выдерживают 7–10 дней в темном месте для того, чтобы прошли все окислительно-восстановительные процессы с примесями, содержащимися в воде. Затем раствор фильтруют. Только после этого концентрация раствора становится постоянной и его можно стандартизировать по щавелевой кислоте или по оксалату аммония. Растворы KMnO4 следует хранить в бутылях из темного стекла. Приготовленный таким способом раствор перманганата калия с молярной концентрацией эквивалента 0,05 моль/л и выше не изменяет свой титр довольно продолжительное время.
Стандартизация раствора перманганата калия по щавелевой кислоте или оксалату аммония (натрия)
Способ определения основан на окислении щавелевой кислоты перманганат-ионами в кислой среде:
5H2C2O4 + 2KMnO4 + 3H2SO4 → 2MnSO4 + 10CO2 + K2SO4 + 8H2O
При этом полуреакции окисления и восстановления имеют вид:
H2C2O4 – 2e— = 2CO2 + 2H+
MnO2— + 8H+ + 5e— = Mn2+ + 4H2O
Тогда fэкв (H2C2O4) = 1/2; fэкв (KMnO4) = 1/5
При комнатной температуре эта реакция протекает медленно. И даже при повышенной температуре скорость ее невелика, если она не катализирована ионами марганца(II). Нагревать кислоту выше 70-80 °С нельзя, так как при этом часть кислоты окисляется кислородом воздуха:
H2C2O4 + O2 → 2CO2 + H2O2
2H2O2 → 2H2O + O2
Реакция взаимодействия перманганата калия со щавелевой кислотой относится к автокаталитическим реакциям. Реакция окисления щавелевой кислоты протекает в несколько стадий. Первые капли перманганата калия даже в горячем растворе обесцвечиваются очень медленно. Для ее начала необходимо присутствие в растворе хотя бы следов Mn2+:
1. MnO4— + MnC+O4 → MnO42- + MnC2O4+
Образовавшийся манганат-ион MnO42- в кислом растворе быстро диспропорционирует:
2. Mn (VI) + Mn (II) = 2 Mn (IV)
3. Mn (IV) + Mn(II) = 2Mn (III)
Марганец (III) образует оксалатные комплексы состава [Mn(C2O4) n] (3-2n)+, где n = 1,2,3; эти комплексы медленно разлагаются с образованием Mn2+ и CO2
Таким образом, пока в растворе не накопится в достаточных концентрациях марганец (II), реакция между MnO4— и С2O42- протекает медленно. Когда концентрация марганца(II) достигает определенной величины, реакция начинает протекать с большой скоростью.
Интенсивная окраска раствора перманганата калия осложняет измерение объемов титранта в бюретке. На практике удобно за уровень отсчета принимать поверхность жидкости, а не нижнюю часть мениска.
Оксалат аммония обладает некоторыми преимуществами по сравнению с другими установочными веществами:
— хорошо кристаллизуется и легко растворяется в воде,
— имеет определенный химический состав и не изменяется при хранении,
— не взаимодействует с кислородом воздуха и СО2 .
Для установки концентрации (титра или молярной концентрации эквивалентов) стандартного раствора перманганата калия рассчитывают навеску щавелевой кислоты H2С2O4 · 2H2O или оксалата аммония (NH4)2C2O4, необходимую для приготовления раствора с молярной концентрацией эквивалента 0,1 моль/л (или 0,05 н.). Рассчитанное количество кислоты (или соли) взвешивают на аналитических весах. Взвешенную массу кислоты (или соли) растворяют в воде в мерной колбе, раствор тщательно перемешивают. Затем титруют раствор KMnO4. Расчет концентрации перманганата калия во всех случаях проводят на основании закона эквивалентов:
N (1/2 H2C2O4) = n (1/5 KMnO4)
Тогда C (1/2 H2C2O4 · 2H2O) · V (H2C2O4 · 2H2O) = C (1/5 KMnO4) · V (KMnO4)
Поскольку ион MnO—4 является сильным окислителем (особенно в кислой среде), то метод перманганатометрии применяется для определения различных восстановителей таких как: H2O2, Fe2+, NO—2, некоторых органических веществ.
Выполнение эксперимента
В коническую колбу для титрования наливают мерным цилиндром 20 мл раствора H2SO4 (1:5) и нагревают до 80 – 90 °С.
Бюретку ополаскивают раствором KMnO4, доводят уровень жидкости до нулевой отметки по верхней границе раствора. Проверяют, нет ли воздушного пузыря в носике бюретки.
В горячий раствор кислоты пипеткой вносят 10,00 мл раствора щавелевой кислоты (оксалата натрия) и титруют раствор кислоты раствором перманганата калия. В начале титрования прибавляют раствор KMnO4 из бюретки по 0,5 мл, дожидаясь обесцвечивания раствора в колбе, перемешивая раствор кругообразными движениями. Для лучшего определения окраски под колбу помещают лист белой бумаги. Когда обесцвечивание раствора вследствие автокатализа будет проходить быстро, раствор титранта прибавляют по каплям. В точке эквивалентности от прибавления одной капли титранта раствор приобретает бледно-розовую окраску устойчивую в течение 30 секунд. Оттитровывают еще две пробы по той же методике. Результаты параллельных определений не должны отличаться более чем на 0,1 – 0,2 мл. Рассчитывают концентрацию и титр определяемого вещества в растворе, используя среднее значение объема титранта, пошедшего на титрование.
Определение железа (II)
Титрование железа (II) основано на реакции:
10FeSO4 + 2KMnO4 + 8H2SO4 = 5 Fe2 (SO4) 3 + 2MnSO4 + K2SO4 + 8H2O
Титровать железо(II) перманганатом калия можно в сернокислой или солянокислой средах. В первом случае не наблюдается никаких осложнений. Присутствие в титруемом растворе хлорид-ионов приводит к перерасходу перманганата и получению нечеткого конца титрования. Это вызвано тем, что реакция между железом(II) и перманганатом индуцирует реакцию между ионами MnO4— и Cl—
MnO4— + 10Cl— + 16H+ → 2Mn2+ + 5Cl2 + 8H2O
Причем в отсутствие ионов Fe2+ эта реакция не идет. Реакции подобного типа, не идущие одна без другой, Н.А. Шилов назвал сопряженными или индуцированными. Индуцированной реакции не возникает, если в растворе присутствуют в достаточных количествах фосфорная кислота и марганец (II). Поэтому перед титрованием в раствор добавляют смесь Рейнгарда-Циммермана, состоящую из серной, фосфорной кислот и сульфата марганца(II). Присутствие H2SO4 в этой смеси создает требуемую концентрацию протонов в титруемом растворе. Присутствие H2PO4 необходимо для связывания железа (III) в бесцветный комплекс и образования фосфатных комплексов марганца (III). Если железо не маскировать, то окраска его комплексных хлоридов будет затруднять наблюдение бледно-розовой окраски в конце титрования перманганатом калия. Железо(III) перед титрованием необходимо восстановить до железа(II).
Выполнение эксперимента. Раствор или навеску анализируемого вещества помещают в мерную колбу вместимостью 100 мл (Vk), добавляют примерно 50 мл 2 н. раствора серной кислоты и водой доводят объем содержимого колбы до метки, тщательно перемешивают. Аликвотную часть полученного раствора (Va 10,00 мл) осторожно переносят в коническую колбу для титрования вместимостью 250 мл, добавляют 5 мл смеси Рейнгарда-Циммермана, 100 мл воды и при интенсивном перемешивании медленно оттитровывают раствором перманганата калия до появления бледно-розовой окраски устойчивой в течение 30 с. Отмечают объем израсходованного титранта (V1). Оттитровывают еще две пробы по той же методике. Результаты параллельных определений не должны отличаться более чем на 0,1-0,2 мл. Вычисляют содержание железа (II) в анализируемом растворе или навеске исследуемого вещества:
m(Fe) = C (1/5 KMnO4) · V (KMnO4) · M(Fe) · (Vk/Va)

Схемы основных реакций, используемых в Red-Ox-метрии

1. Окислительно — восстановительный эквивалент
Пример. Вычислить эквивалент дихромата калия K2Cr2O7 в реакции взаимодействия с йодидом калия KI в кислой среде.
Решение:
1-й способ (по изменению степени окисления)
В реакции восстановления иодидом ион Cr2O72- восстанавливается до иона Cr3+. Схема реакции:
K2Cr2O7 + KI + H2SO4 → Cr2(SO4)3 + I2 + K2SO4 + H2O.
Изменение степени окисления хрома равно 6, что видно из электронного уравнения, записанного для процесса восстановления:
2Cr+6 + 6e— = 2Cr3+,
ЭK2Cr2O7 = М (K2Cr2O7) / 6 = 294,22 / 6 = 49,04
2-й способ (по электронно – ионному уравнению)
Составляем электронно – ионное уравнение для процесса восстановления:
Cr2O72- + 14H+ + 6e— = 2Cr3+ + 7H2O,
ЭK2Cr2O7 = М (K2Cr2O7) / 6 = 294,22 / 6 = 49,04
Последовательность составления электронно-ионных уравнений следующая:
1. Записывают схему полуреакции в ионном виде:
Cr2O72- + H+ → 2Cr3+ + H2O
2. Проверяют число атомов каждого элемента в левой и правой частях уравнения (должно соблюдаться их равенство):
Cr2O72- + 14H+ → 2Cr3+ + 7H2O
3. Уравнивают суммы зарядов. Сумма и знак электрических зарядов в левой и правой частях уравнения должны совпадать:
Cr2O72- + 14H+ + 6e— = 2Cr3+ + 7H2O.
Так же составляют электронно – ионное уравнение для процесса окисления иодид-ионов:
2I— — 2e— = I2.
Исходя из правила, по которому число электронов, отданных восстановителем, должно быть равно числу электронов, полученных окислителем, умножают электронно – ионные уравнения на соответствующие множители.
Путем суммирования электронно – ионных уравнений для процессов восстановления и окисления, умноженных на соответствующие коэффициенты, получают ионное уравнение реакции, по которому составляют молекулярное уравнение:
Cr2O72- + 6I— + 14H+ = 3I2 + 2Cr3+ + 7H2O
K2Cr2O7 + 6KI + 7H2SO4 = 3I2 + Cr2(SO4)3 + 4K2SO4 + 7H2O.
При составлении электронно-ионных уравнений нужно руководствоваться следующим:
- Если продукт реакции содержит меньше кислорода, чем исходное вещество, то в кислой среде происходит связывание кислорода и ионов водорода с образованием воды. В нейтральной и щелочной средах освобождающийся кислород взаимодействует с водой, образуя гидроксильные ионы.
- Если продукт реакции содержит больше кислорода, чем исходное вещество, то в нейтральных и кислых растворах расходуется вода, а в щелочных растворах – ионы гидроксила.
2. Йодометрия
Пример 1. К навеске 0,1275 г дихромата калия K2Cr2O7 прибавлен избыток иодида калия; выделившийся иод оттитрован 22,85 мл раствора тиосульфата натрия Na2S2O3∙5H2O. Найти нормальность тиосульфата и его титр по иоду.
Решение.
K2Cr2O7+6KI+7H2SO4=4K2SO4+Cr2(SO4)3+3I2+7H2O
Cr2O72- + 14H+ + 6e— = 2Cr3+ + 7H2O,
2I— — 2e— = I2,
ЭK2Cr2O7=М / 6 =49,03, ЭI2 =М /2 126,9.
Определяем количество миллиграмм-эквивалентов дихромата калия в навеске 0,1275 г:
127,5 / 49,03 = 2,6 (мг-экв).
Столько же миллиграмм-эквивалентов тиосульфата натрия Na2S2O3∙5H2O прореагировало с дихроматом калия.
Нормальность тиосульфата натрия равна:
2,6 / 22,85 = 0,1142
Титр тиосульфата натрия по йоду равен:
ТNa2S2O3∙5H2O/I2 = (Сн (Na2S2O3∙5H2O) * Мэ (I2)) / 1000
ТNa2S2O3∙5H2O/I2 = (0,11452*126,9) / 1000 = 0,014480 (г—мл).
Пример 2. Определить количество сульфата натрия в исследуемом растворе, если на титрование I2, выделенного из иодида калия эквивалентным ионам SO42- количеством ионов CrO42-, затрачено 12,00 мл 0,0540 н. раствора тиосульфата натрия Na2S2O3∙5H2O.
При йодометрическом определении сульфат-ионы SO42- замещаются эквивалентным количеством хромат ионов, которые определяются иодометрически:
Na2SO4 + BaCrO4 → ↓BaSO4 + Na2CrO4.
Избыток хромата и сульфата бария удаляют путем фильтрования. Количество хромата натрия Na2CrO4, эквивалентное количеству сульфата натрия Na2SO4, определяют иодометрически:
2Na2CrO4+6KI+8H2SO4=3I2+Cr2(SO4)3+2Na2SO4+8H2O+3K2SO4,
CrO42- + 8H+ + 3e— = Cr3+ + 4H2O, ЭNa2CrO4 = .
Согласно уравнению реакции молекуле сульфата натрия соответствует одна молекула хромата натрия. Следовательно,
ЭNa2SO4=М /3 = 142,048 / 3 = 47,349.
Количество миллиграмм-эквивалентов хромата натрия равно:
N(Na2S2O3)∙V(Na2S2O3)=12∙0,0540=0,648 (мг-экв).
Количество сульфата натрия в растворе равно:
0,648∙47,35=30,68 (мг) = 0,0307 (г).
3. Перманганатометрия
Пример 1. К 100 мл исследуемой воды прибавлено 10,00 мл 0,0120 н. раствора перманганата калия и 5,00 мл серной кислоты (1:3).
После кипячения в течение 10 мин добавлено 10,00 мл 0,0120 н. раствора щавелевой кислоты H2C2O4∙2H2O, избыток которой оттитрован, 4,50 мл 0,0102 н. раствора перманганата калия. Определить окисляемость воды.
Решение. 1-й способ
Окисляемость воды определяется количеством миллиграммов перманганата калия или кислорода, затраченных на окисление примесей в 1 л воды. Используется метод обратного титрования.
Общее количество прибавленного раствора перманганата калия равно:
10,00+4,50=14,50 (мл).
Количество миллиграмм-эквивалентов перманганата калия равно:
VKMnO4∙NKMnO4 = 14,50∙0,0102=0,1479 (мг-экв).
Количество миллиграмм-эквивалентов щавелевой кислоты H2C2O4∙2H2O равно:
V(H2C2O4∙2H2O) ∙N(H2C2O4∙2H2O)=10,00∙0,0120=0,1200 (мг-экв).
Щавелевая кислота взаимодействует с перманганатом калия, не вступившим в реакцию с примесями воды.
Количество перманганата калия, затраченное на окисление примесей, содержащихся в 100 мл воды, равно:
0,1479-0,1200=0,0279 (мг-экв).
Умножив количество миллиграмм-эквивалентов на эквивалент перманганата калия или кислорода, определяем окисляемость воды, выраженную в миллиграммах перманганата калия или кислорода. ЭKMnO4=31,6, ЭO2=8,0.
0,0279∙10,00∙31,6=8,82 (мг KMnO4),
или
0,0279∙10,00∙8,0=2,232 (мг О2).
2-й способ
Общий объем раствора перманганата калия равен:
VKMnO4=10,00+4,50=14,50 (мл).
Объем раствора KMnO4, вступивший в реакцию со щавелевой кислотой, равен:
VKMnO4 = (V(H2C2O4∙2H2O) *N(H2C2O4∙2H2O)) / N (KMnO4),
VKMnO4 = (10,00 * 0,0120) / 0,0120 =11,76 (мл).
На окисление примесей в 100 мл воды затрачено перманганата калия:
14,50-11,76=2,74 (мл).
Окисляемость воды по KMnO4 равна:
2,74∙10,00∙31,6∙0,0102=8,83 (мг KMnO4).
Окисляемость воды по О2 равна:
2,74∙10,00∙8,0∙0,0102=2,236 (мг О2).
Метод перманганатометрии широко используется при проведении технических анализов.
Пример 2. Определить процентное содержание оксида марганца (IV) в загрязненном пиролюзите, если образец его 0,1530 г обработан 30,00 мл 0,1075 н. раствора щавелевой кислоты и разбавленной серной кислотой. Для титрования избытка щавелевой кислоты требуется 5,31 мл 0,1100 н. раствора перманганата калия.
Решение. 1-й способ
MnO2+H2C2O4+H2SO4=MnSO4+2H2O+2CO2↑,
MnO2+4H++2e−=Mn2++2H2O,
С2O42- — 2e− = 2CO2↑,
ЭMnO2=М / 2=86,94 / 2 .
Объем избытка раствора щавелевой кислоты равен:
V(H2C2O4∙2H2O)=(V(KMnO4) * N(KMnO4)) / N(H2C2O4∙2H2O)
V(H2C2O4∙2H2O)= (5,31 * 0,1100) / 0,1075 = 5,44 (мл).
Количество щавелевой кислоты, израсходованной на обработку навески, равно:
30,00-5,44=24,56 (мл).
Количество миллиграмм-эквивалентов щавелевой кислоты H2C2O4∙2H2O:
24,560,1075=2,64 (мг-экв).
Количество оксида марганца (IV), эквивалентное количеству щавелевой кислоты H2C2O4∙2H2O:
2,64∙43,47 = 114,75 (мг) = 0,1147 (г),
%MnO2 =(0,1147 * 100) / 0,1530 =74,97.
2-й способ
Количество миллиграмм-эквивалентов щавелевой кислоты H2C2O4∙2H2O:
30,00∙0,1075=3,2250 (мг-экв).
Количество миллиграмм-эквивалентов щавелевой кислоты H2C2O4∙2H2O, находящееся в избытке:
5,31∙0,1100=0,5841 (мг-экв).
Количество миллиграмм-эквивалентов щавелевой кислоты H2C2O4∙2H2O, вступившее в реакцию с оксидом марганца (IV):
3,2250-0,5841≈2,64 (мг-экв).
Количество миллиграммов оксида марганца (IV):
2,64∙43,47=114,75 (мг) = 0,1147 (г),
%MnO2 = (0,1147*100)/0,1530=74,97.
4. Кривые окислительно – восстановительного титрования
В методах окисления – восстановления большое значение имеет вопрос о правильности выбора обратимого окислительно-восстановительного индикатора и построения кривых титрования.
В процессе окислительно – восстановительного титрования величина потенциала изменяется. Наглядно процесс титрования изображается в виде кривых, где величина потенциала представлена как функция объема титрующего раствора.
Пример 1.
Построение кривой титрования и ее разбор рассмотрим на примере реакции
Fe2+ + Ce4+ ↔ Fe3+ + Ce3+,
которая протекает в прямом направлении.
EoFe3+/Fe2+= +0,77 в, EoCe4+/Ce3+= 1,44 в.
Концентрации электролитов одинаковы, первоначальный объем раствора равен 100 мл.
Титруем раствор соли закисного железа солью церия. Во всех точках, кроме начальной, существуют две окислительно – восстановительные системы: Fe3+/Fe2+ и Ce4+/Ce3+. При добавлении каждой порции титрующего раствора в процессе установившегося равновесия потенциалы обеих систем становятся равными. Для вычисления величин потенциалов в различные моменты титрования можно пользоваться обоими уравнениями полуреакций, т. е.
Fe2+ — e−↔ Fe3+,
Ce4+ + e− ↔ Ce3+.
В области до точки эквивалентности потенциал удобно вычислять по концентрации окисленной и восстановленной форм титруемого раствора, т.е. по концентрации окисленной и восстановленной форм системы Fe3+/Fe2+.
Рассчитываем значение потенциала для разных моментов титрования.
1. К 100 мл 0,1 н. раствора соли железа (II) прибавлено 50 мл соли церия (IV). Растворы одинаковых нормальных концентраций реагируют в равных объемах.
[Fe3+]/[Fe2+] = 50 / 50 = 1, [Fe3+] = [Fe2+]
E = EoFe3+/Fe2+ + 0,059lg ([Fe3+]/[Fe2+]) = 0,77 (в).
2. При добавлении 99 мл соли Ce4+ к 100 мл 0,1 н. раствора соли железа отношение концентраций
[Fe3+]/[Fe2+] = 99 / 1 = 99,
значение потенциала:
E = 0,77+0,059lg99 = 0,89 (в).
3. Прилитый объем соли Ce4+ равен 99,9 мл.
E = 0,77+0,059lg (99/0.1) = 0,77 + 0,059 lg 990 = 0,95 (в).
4. Прилито 100 мл соли церия.
Значение потенциала в точке эквивалентности определяют из уравнения:
Еэкв = (n1*E10 + n2*E20) / (n1+ n2)
где n1 и n2 – стехиометрические множители;
и – стандартные окислительно-восстановительные потенциалы соответствующих пар.
Еэкв = (1,44 + 0,77) / 2 = 1,105 (в).
После достижения точки эквивалентности соотношение концентраций окисленной и восстановленной форм находят по объему титрующего раствора, т. е. определяют значение потенциала пары Ce4+/Ce3+ в различные моменты титрования.
5. Объем прибавленной соли Ce4+ равен 100,1 мл (избыток). Потенциал системы:
E = Eo + 0,059lg( [Ce4+] / [Ce3+]),
E = 1,44 + 0,059lg (0,1/100) = 1,44 + 0,059 * lg10-3= 1,263 (в).
Величина скачка ∆Е потенциала около точки эквивалентности определяется 0,1 мл недостатка и 0,1 мл избытка титрующего раствора по отношению к эквивалентному объему и лежит в пределах 0,95 в – 1,263 в.
Область скачка можно расширить, если один из образующихся при реакции ионов связывать в комплекс.
При добавлении 200 мл окислителя [Ce4+] =[ Ce3+], отсюда
E = Eo = 1,44 в.
Пример 2. На сколько процентов оттитрован раствор, если к 20,00 мл 0,1 н. раствора сульфата железа (II) прибавляли 0,1 н. раствор сульфата церия (IV) до момента, когда потенциал стал равен 0,92 в?
Решение
Fe2+ + Ce4+ ↔ Fe3+ + Ce3+
EoFe3+/Fe2+= +0,77 в, EoCe4+/Ce3+= 1,44 в.
Определяем значение потенциала в точке эквивалентности:
Еэкв = (0,77 + 1,44) / 2 = 1,105 (в).
Так как величина потенциала раствора в данный момент титрования меньше 1,105 в, рассчитываем концентрацию не оттитрованной соли железа по уравнению
E = EoFe3+/Fe2+ + 0,059lg ([Fe3+] / [Fe2+]).
Допустим, что в данный момент оттитровано x мл соли железа (II); соответственно образовалось x мл соли железа (III), осталось неоттитрованным 20-х (мл) соли железа (II).
Отношение равновесных концентраций окисленной и восстановленной форм выражаем в виде отношения объемов:
0,92=0,77+0,059lg (х/ (20-х)), где 0,059lg (х/ (20-х)) = 0,15,
lg (х/ (20-х)) = 2,54, (х/ (20-х)) = 346,7, x = 6934-346,7х,
347,7х=6934, х=19,94.
Таким образом, оттитровано 19,94 мл соли железа (II), что составляет 99,70% от 20,00 мл. Не оттитровано 0,06 мл раствора сульфата железа (II) или 0,3%.
5. Направление реакций окисления – восстановления
Пример 1. В какую сторону будет направлена реакция, выражаемая уравнением:
AsO43- + 2I— + 2H+ ↔ AsO33- + H2O + I2
При pH=7 и равной концентрации окислителя и восстановителя?
Решение:
Записываем величины стандартных окислительно-восстановительных потенциалов систем и уравнения полуреакций:
EoAsO43-/AsO33-= +0,56 в, EoI2/2I—= + 0,53 в,
AsO43- + 2H+ + 2e— ↔ AsO33- + H2O,
2I— — 2e— ↔ I2.
Из уравнений полуреакций видно, что концентрация ионов H+ влияет на величину потенциала системы AsO43-/ AsO33-, потенциал системы I2/2I— не зависит от концентрации ионов H+.
EAsO43-/AsO33- = EoAsO43-/AsO33- + (0,059 / 2) * lg (([AsO43-] * [10-7]2) / [AsO33-]),
EAsO43-/AsO33- = 0,56 +0,0295 * lg (10-7)2 0,56 — 0,059=0,147 (в),
Еокисл – Евосст = ЕI2/2I— — EAsO43-/AsO33- = 0,536-0,147=0,389 (в).
При pH=7 потенциал системы I2/2I— больше потенциала системы
AsO43-/AsO33-, поэтому наиболее сильным окислителем будет I2; направление реакции смещается в сторону окисления ионов AsO33- в ионы AsO43-.
Пример 2. В какую сторону будет направлена рассмотренная в предыдущем примере реакция при ph = 0? [H+] = 1 г-ион/л.
Решение:
EAsO43-/AsO33- = 0,56 + (0,059 / 2) * lg1 = 0,56 (в),
∆E = EAsO43-/AsO33- — ЕI2/2I— = 0,56-0,53=0,03 (в).
Реакция идет в направлении:
AsO43- + 2I— + 2H+ → AsO33- + H2O + I2.
Константа равновесия этой реакции невелика, поэтому при создании необходимых условий ее можно смещать в желаемом направлении.
K = ([AsO33-]*[ I2]) / ([AsO43-]*[ I—]2 *[H+]2)
lgK = ((0,56 – 0,53) * 2) / 0,059 = 1.
Отсюда К = 10.
При увеличении концентрации ионов H+ или AsO43- повышается значение потенциала пары AsO43-/AsO33- и реакция протекает в прямом направлении. Наоборот, при уменьшении концентрации ионов H+ равновесие реакции смещается в обратную сторону.
Когда значение К велико, реакция является практически необратимой. Например, для реакции
Sn2+ + 2Fe3+↔2Fe2+ + Sn4+,
lgK = ((0,77-0,15) * 2) / 0,059 ≈21.
Отсюда
K = ([Fe2+]2*[ Sn4+]) / ([Fe3+]2*[ Sn2+]) = 1021
Реакция в прямом направлении практически идет до конца.