1146 просмотров
Здравствуйте, ребёнку 3 недели, сегодня позвонили из поликлиники,повышение итр(подозрение на муковисцидоз) в 2 раза!!!норма 65,у нас 130!!!я очень переживаю, такое повышение говорит о диагнозе???сегодня взяли повторный анализ крови,пока будет результат, я вся изведусь…Ребёнок на гв,стул частый,после каждого кормления,желто зелёный без запаха сильного,не срыгивает,начиталась, теперь и лоб кажется солоноватый у ребенка…набрал за 3 нед 500 грамм.
На сервисе СпросиВрача доступна бесплатная консультация педиатра онлайн по любой волнующей Вас проблеме. Врачи-эксперты оказывают консультации круглосуточно. Задайте свой вопрос и получите ответ сразу же!

Педиатр
Здравствуйте, нужно набраться терпения , перепроверить ещё раз. Прибавка впринципе нормальная для 3х недель. Стул обильный?
Наталья, 5 ноября 2022
Клиент
Ольга, он после каждого кормления, в среднем около наверное чайной ложки,но у меня и со страшим ребёнком в первые месяцы на гв такой частый стул бвл,поэтому как то не запозртла ничего. СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
На гв это норма. Вообще при муковисцидозе стул обильный и ребёнок плохо прибавляет в весе. На моей практике был такой случай, когда не подтвердили МВ да.

Педиатр, Дерматолог, Венеролог, Детский
Здравствуйте, Наталья
В данном случае не исключена ошибка, но в любом случае нужно пересдать и ждать результата. Достаточно часто бывает дефект сбора, поэтому раньше времени не переживайте
Наталья, 5 ноября 2022
Клиент
Дарья Николаевна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр, Дерматолог, Венеролог, Детский
Да, на моей практике было.
Не очень часто, пару раз точно было

Педиатр
Здравствуйте, нужно дождаться повторного результата, терпения🙏🏻Стул такой частоты, консистенции и цвета в норме у всех младенцев
Наталья, 5 ноября 2022
Клиент
Алина Александровна, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Педиатр
Да, может, бывают ошибки лаборатории. Про частоту таких случаев однозначно нельзя сказать, но раньше времени не переживайте, малыш чувствует беспокойство мамы и тоже может беспокоиться

Педиатр, Нейрохирург
Здравствуйте!
Делать выводы пока ещё рано. Поэтому и сделали повторный забор крови, чтобы исключить неправильный забор и лабораторные ошибки.
Самое главное сейчас успокоиться и ждать результата. Будем надеяться, что это ошибка.
Стул пенится?
Наталья, 5 ноября 2022
Клиент
Эндже, СКАЖИТЕ пожалуйста на вашей практике такое повышение может дать ложноположит.результат и часто ли они?
Стул не пенится
Педиатр, Нейрохирург
Положительный результат скрининга говорит о том, что есть подозрение на муковисцидоз.
Это ещё не означает 100% наличие. Часто бывают каждых ложноположительные результаты, 3-6 случаев положительного результата только один оказывается действительно больным муковисцидозом, а оставшиеся — это ложноположительные результаты.
В моей практике ещё не было положительных результатов.
Обычно, для постановки диагноза решающее значение имеет потовый тест,

Педиатр
Здравствуйте) нужно дождаться повторного результата, в лабораториях бывают ошибки.

Педиатр
Здравствуйте Наталья, судя по описанию абсолютно здоровый малыш, набор в весе хороший , надеюсь результат ошибочный, наберитесь терпения не переживайте, будем ждать повторный результат, как придут отпишитесь. Будьте здоровы!
Оцените, насколько были полезны ответы врачей
Проголосовало 0 человек,
средняя оценка 0
Что делать, если я не нашел ответ на свой вопрос?
Если у Вас похожий или аналогичный вопрос, но Вы не нашли на него ответ — получите свою онлайн консультацию врача.
Если Вы хотите получить более подробную консультацию врача и решить проблему быстро и индивидуально — задайте платный вопрос в приватном личном сообщении. Будьте здоровы!
Introduction
Cystic fibrosis (CF) is an autosomal recessive genetic disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene (1). Early identification of individuals with CF is critical towards improved growth and development, as well as survival (1, 2). Implementation of newborn screening (NBS) nationally has led to earlier detection of patients with CF and better outcomes for this patient population (3).
NBS programs across the world use a variety of algorithms for screening. Most begin with the measurement of immunoreactive trypsinogen (IRT), which is produced by the pancreas and elevated in the blood of infants with CF (4). In Alabama, newborns with an elevated IRT on NBS will then obtain a screening CFTR gene mutation analysis using a commercially available panel (5, 6). In general, newborns with an elevated IRT and at least one mutation identified on the genetics panel are considered to have a positive NBS and are immediately referred for confirmatory testing with a sweat chloride test (SCT). An infant with a normal sweat chloride (SC) concentration <30 mmol/L and only one mutation is designated a carrier, whereas an infant with a SC concentration ≥ 60 mmol/L and/or two known CF disease-causing mutations confirms a definitive diagnosis of CF (7).
Advent of the NBS has led to the identification of asymptomatic infants found to have a positive NBS with at least one mutation and a SC concentration between 30 and 59 mmol/L, who are considered to have an inconclusive diagnosis and may be designated to have CF Screen Positive, Inconclusive Diagnosis (CFSPID), also known as CFTR-related metabolic syndrome (CRMS) (8–15). Repeated SCT in the diagnostic range or identification of two known CF disease-causing mutations can subsequently confirm the diagnosis of CF (15, 16). Some children do not develop diagnostic range SCT (14–17). Children may also present with one or more mutations in CFTR of variable clinical consequence (VCC), which may result in symptomatic CF but are also found in healthy individuals who do not develop any symptoms of disease (5, 7). Typically, children who carry the designation of CRMS remain free of symptoms for years and may never develop symptoms consistent with a CF diagnosis. However, several longitudinal studies have noted anywhere from 5% to 48% of these children will develop clinical features suggestive of CFTR-related disorder (CFTR-RD) or CF (5, 7, 13, 16–20). Despite this notable risk of transition, there are no clear predictive biomarkers to identify which children with CRMS have a higher risk of latter transitioning to a diagnosis of CF disease. A few global studies have demonstrated a potential association between initial IRT values and genetics that predict transition to a CF diagnosis (21–24). Two retrospective studies noted a higher likelihood of transition to CF if an infant’s initial SCT value was between 50 and 59 mmol/L (15) or if SCT progressed toward diagnostic range faster (24). These biochemical markers are not yet part of guidelines for CRMS management and are challenging to use on an individual basis to counsel families. Furthermore, current guidelines remain unclear on the frequency and duration of follow-up for patients with CRMS (16). In this study, we report the outcomes of children with CRMS after implementation of NBS in the only accredited CF center in the state, to further add to the current literature on this topic and explore novel approaches to next steps for guideline development.
Methodology
State protocol and confirmatory testing
The state of Alabama uses an IRT/DNA protocol that involves a 2-step process of measuring IRT followed by screening for cystic fibrosis transmembrane regulator (CFTR) mutations on a limited DNA panel for any infants with an IRT value >96th percentile on that designated day (6). The NBS is considered positive if an infant has IRT value >96th percentile AND carries at least 1 mutation identified on the DNA screening panel or if the infant has an IRT value >99.9th percentile for the day. The Alabama Department of Public Health used InPlex® CF Molecular Test (Hologic, Ltd.) until August 2016, then changed to the xTAG® Cystic Fibrosis (CFTR) 39 v2 K (Luminex Corporation, Austin, Texas, United States.) A small number of newborns are referred for other reasons such as a very high IRT/failed NBS, family history of CF, or other reasons for concern. Any NBS-positive infants then undergo confirmatory SCT at our CF-accredited Center here at the University of Alabama at Birmingham (UAB) within Children’s of Alabama (CoA) Hospital. Additional genetic analysis (CFTR sequencing with deletion/duplication analysis) was performed at provider discretion (persistently intermediate SCT or heightened clinical suspicion). The combination of the information provided by the NBS and confirmatory SCT provides the infant with their initial classification (per the most recently updated guidelines (13)) of one of the following: non-carrier (SCT <30 mmol/L and no CFTR mutations), carrier status (SCT <30 mmol/L and one CFTR mutation), CF (SCT ≥60 mmol/L and/or two disease-causing mutations), or CRMS (indeterminate SCT 30–59 mmol/L and less than two disease-causing mutations OR SCT <30 mmol/L and two CFTR mutations with at least one of unclear phenotypic consequences).
Study population
This study was approved by the UAB Institutional Review Board (#300005123). We retrospectively reviewed the charts of all newborns born in Alabama between the years 2008 to 2020 referred to our center for evaluation of cystic fibrosis. Initial newborn screening results, including IRT value and mutation screening results, were collected for each of these infants. Data collected from available charts for any follow-up visits through January 2021 included self-reported race/ethnicity, sex, first quantifiable SCT, CFTR genetic results, microbiology results from sputum swabs, stool elastase, lung function, weight, and height measures. Infants initially designated CRMS were sub-classified by final diagnosis when data collection ended. A child transitioned to carrier status (CRMS-Carrier) with normalized SCT and if only one CFTR mutation identified. If a child met criteria for CF diagnosis [4] with repeat SCT concentration resulted >60 mmol/L, expanded genetics revealed a second CF-causing mutation, development and persistence of clinical symptoms, or with re-classification of their VCC mutation to a CF-causing mutation (determined by CFTR2 project database (25)), then they transitioned to a CF diagnosis (CRMS-CF). All other newborns who remained inconclusive diagnosis remained in the CRMS category as CRMS-Persistent (CRMS-P). Children designated CRMS are followed by pediatric pulmonologists with CF expertise in a separate clinic space than that of children followed in our CF clinics.
Statistical analysis
Descriptive analyses (means, standard deviations, medians, interquartile ranges, frequency distributions (%)) were used to describe patient demographics, clinical characteristics, and outcomes. Results are presented as means +/− standard deviation (SD) or standard error of the mean (SEM) where noted. For all analyses, a p-value of <0.05 was considered statistically significant. We compared initial diagnoses (CF, CRMS, Carrier) and CRMS subgroups (CRMS-CF, CRMS-Carrier, and CRMS-P) using analysis of variance. Tukey’s adjustment for multiple comparisons was completed for each comparison. Anthropomorphic trajectories and SCT trajectories are modeled using multilevel linear mixed models with a random intercept. Change over time is modeled as age. Statistical analysis was performed using STATA software (StataCorp,College Station, Texas) and GraphPad Prism (San Diego, California).
Results
Demographics of NBS population
From 2008 to 2020, there were 1,411 infants identified through NBS with elevated IRT (IRT+) and referred to our CF Center. Sixty-one infants with no CFTR mutation identified on screening genetics and a normal SCT did not undergo any further evaluation, and were therefore excluded from the study. Four infants had insufficient quantities on all SCT attempts in the record and were excluded from the study. The remaining 1,346 infants with IRT+ and ≥1 CFTR mutation identified (IRT+/DNA+) had at least one SCT result documented. Of these 1,346 infants, 1,170 infants (86.9%) had an initial SC concentration <30 mmol/L, 51 infants (3.8%) had an intermediate SC concentration between 30 and 59 mmol/L and 125 infants (9.3%) had an initial SC concentration ≥ 60 mmol/L (Figure 1).
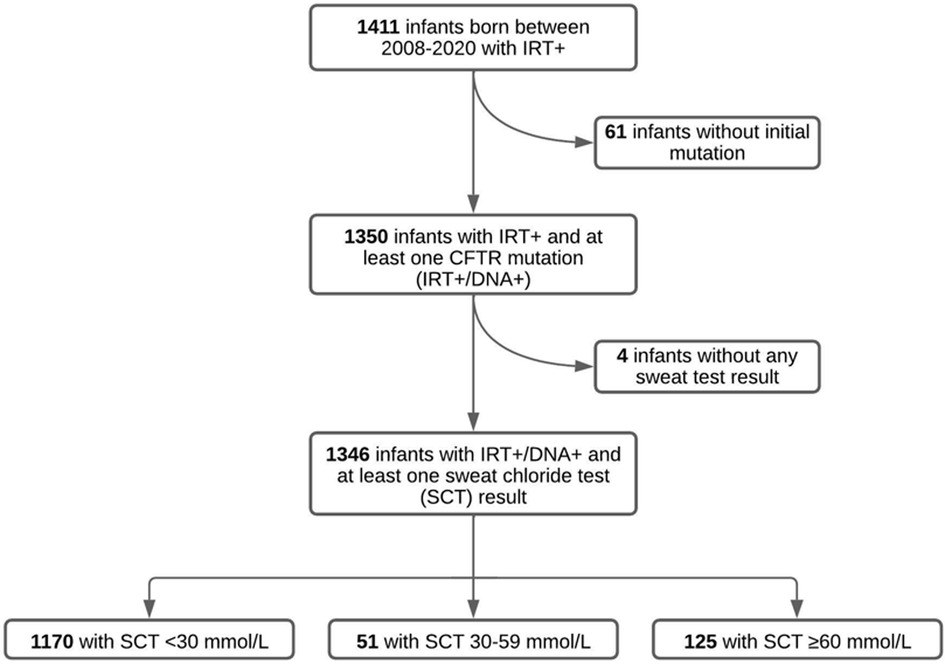
Figure 1. Results of SCT in infants referred for elevated IRT and 1 mutation (IRT+/DNA+) in Alabama between 2008 and 2020. Of the initial 1,411 infants referred for elevated IRT, 65 of them were excluded due to not having an initial CFTR mutation or a documented SCT. Our study population therefore included 1,346 infants with IRT+/DNA+ screening and at least one successful SCT concentration.
Characteristics for all infants who presented with IRT+/DNA+ and confirmatory SCT are shown in Table 1. All 125 infants with SC concentrations >60 mmol/L were classified as CF on initial evaluation. Four infants with initial intermediate SC concentrations had 2 CF-causing mutations result on their NBS DNA mutation panel and were therefore classified as CF on their initial diagnosis. Forty-seven infants with initial intermediate SC concentrations met the criteria for CRMS classification. Of the 1,170 infants with initial SC concentrations <30 mmol/L, 16 resulted with 1 CF-causing mutation and 1 variant of VCC mutation on their NBS DNA panel and were therefore classified as CRMS on initial evaluation. The remaining 1,154 infants with initial SC concentrations <30 mmol/L had only one mutation identified and were classified as Carriers. Therefore, of the total 1,346 infants included in our study analysis, 129 infants (9.6%) were classified as CF on their initial assessment, 63 (4.7%) as CRMS, and 1,154 (86%) as Carriers. This indicates a CF:CRMS ratio of 2.1:1.0. As consistent with known data on IRT in newborns with CF, we found newborns diagnosed with CF after completing the NBS process had a higher IRT and initial SCT value when compared to children initially designated as CRMS (p < 0.001 for IRT; p < 0.0001 for SCT) or Carrier (p < 0.001 for IRT; p < 0.001 for SCT, data not shown). There was also a statistically significant difference seen in children with initial designations of CRMS compared to Carrier for IRT values (p = 0.0281) and first SCT value (p < 0.0001).
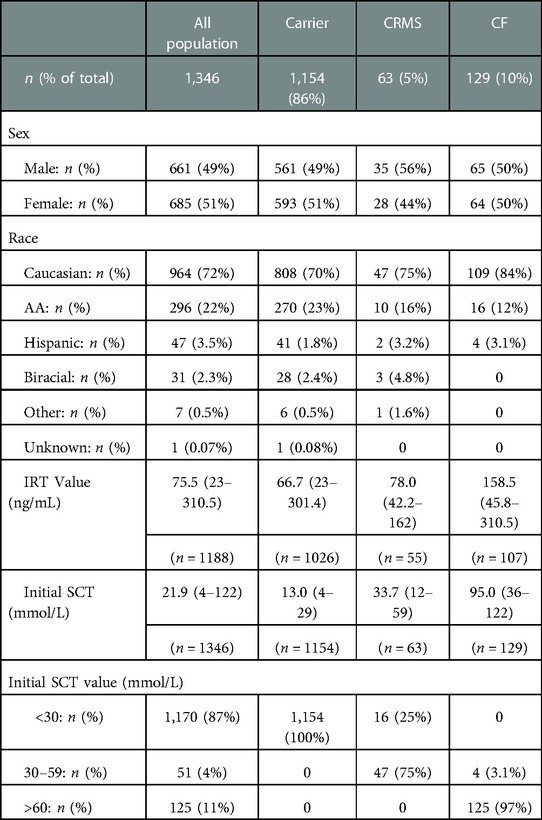
Table 1. Characteristics for all infants with IRT+/DNA+ and at least one successful SCT (n = 1,346).
Outcomes of infants initially designated with CRMS
Characteristics of the 63 infants who were classified as CRMS on their initial encounter are shown in Table 2. Of these 63 infants, 16 (25%) had an initial SC value <30 mmol/L and 47 (78%) had an initial intermediate SC value between 30 and 59 mmol/L. Average IRT values for infants classified as CRMS was 75.2 ng/ml. Within the timeframe of the study, 12 (19%) of the total 63 CRMS infants transitioned to Carrier status (CRMS-Carrier), 40 (64%) of them remained CRMS status given persistent inconclusive diagnosis (CRMS-P), and 11 (18%) of them transitioned to a diagnosis of CF (CRMS-CF). All 12 CRMS-Carrier infants had an initial SC value in the intermediate range. All 12 of these infants had a repeat SCT that normalized to <30 mmol/L. Average IRT values for infants classified as CRMS-Carrier was 67.4 ng/ml. Of the 40 infants who remained CRMS status by the end of the observation period (CRMS-P), 16 (40%) had an initial SC value <30 mmol/L and 24 (60%) had an initial SC value in the intermediate range. Eleven (28%) of these patients were either lost to follow-up or had their care transferred to another CF Center after moving to another state. The remainder 29 (72.5%) patients are followed at our CF center for continued evaluation as per the CRMS guidelines.
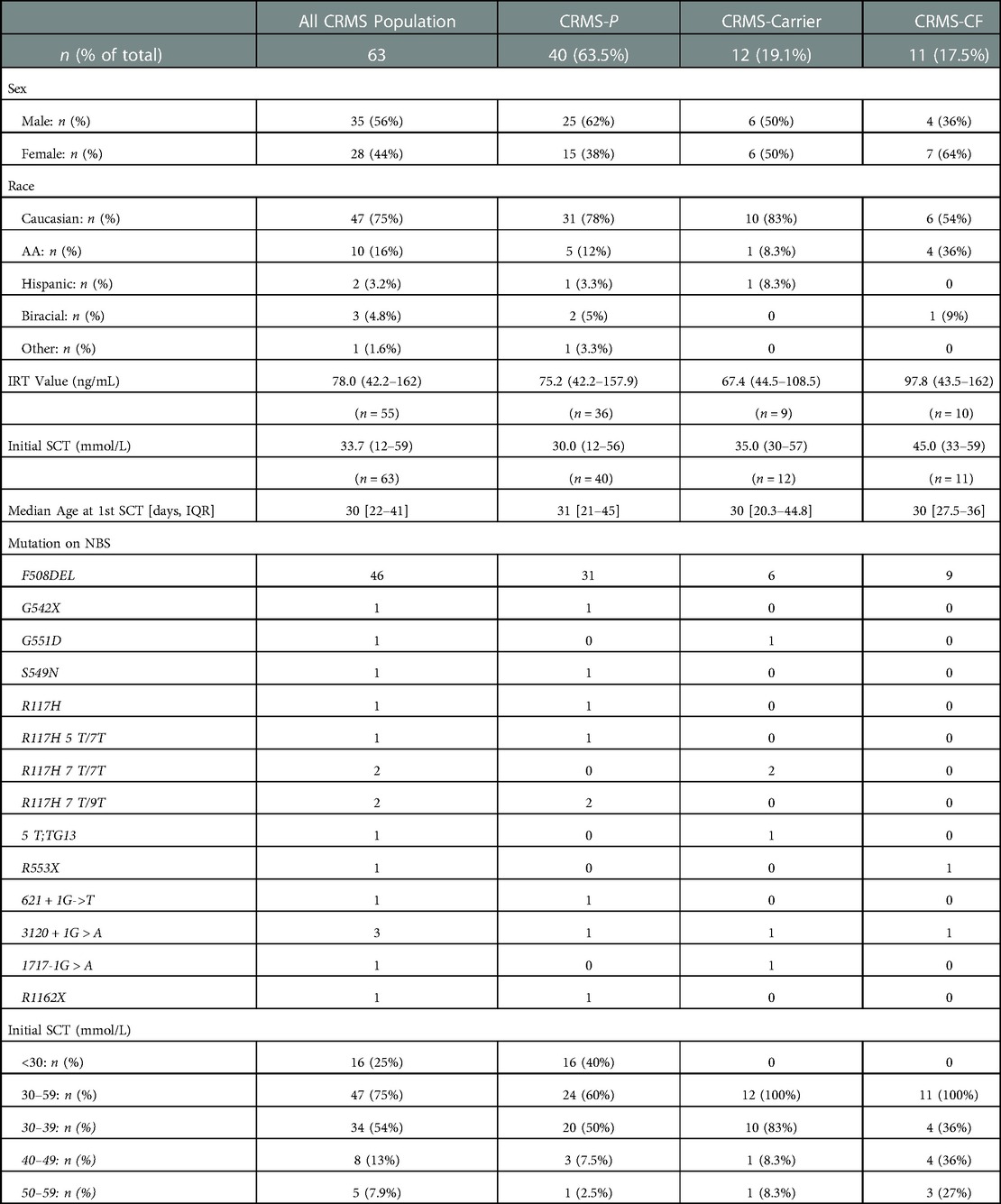
Table 2. CRMS-P, CRMS-Carrier, CRMS-CF diagnosis.
Detailed characteristics of children transitioned from CRMS to cf
Diagnostic data for the 11 total infants initially designated with CRMS who had a final diagnosis of CF (CRMS-CF) are shown in Table 3. All 11 CRMS-CF infants had an initial SC value in the intermediate range. Four (36%) of the 11 CRMS-CF infants had an initial SC concentration between 30 and 39 mmol/L, 4 (36%) had an initial SC concentration between 40 and 49 mmol/L and 3 (27%) had an initial SC concentration between 50 and 59 mmol/L. Five (45%) of them were transitioned to a CF diagnosis after repeat SCT resulted >60 mmol/L, 3 (27%) transitioned after development and persistence of clinical symptoms, 2 (18%) transitioned after expanded genetics revealed a second CF-causing mutation and 1 (9%) was re-classified as CF after one of their mutations was re-classified as CF-causing on the CFTR2 project database. Of the 5 children that transitioned to a CF diagnosis after having repeat SC concentrations result positive, 3 (60%) had initial SC concentrations 50–59 mmol/L, 1 (20%) had initial SC concentrations 40–49 mmol/L, and 1 (20%) had initial SC concentrations 30–39 mmol/L. Of the 3 children that transitioned to CF diagnosis after development of clinical symptoms, 2 (67%) had initial SC concentrations 30–39 mmol/L and 1 (33%) had initial SC concentrations 40–49 mmol/L. All 3 of these children were transitioned to a CF diagnosis due to the persistence of clinical symptoms including cough, spirometry changes, and pulmonary exacerbations that were accompanied by persistent methicillin-resistant staphylococcus aureus (MRSA), pseudomonas aeruginosa or non-tuberculous mycobacteria (NTM) culture growth. These 3 children have also demonstrated upward trending SC values on repeat testing. Of the 2 children that transitioned to a diagnosis of CF after expanded genetics workup revealed a second CF-causing mutation, both had an initial SC concentrations 30–39 mmol/L. The 1 infant who transitioned to CF after their mutation (F191V) was classified as CF-causing had an initial SC concentration 40–49 mmol/L. Average IRT for the 11 children with CRMS-CF was 97.8 ng/ml. The average age for these children when they were transitioned to a diagnosis of CF was 3.9 years of age (range 0.18 years of age to 8.4 years of age). Four of these children were transitioned to a diagnosis of CF after the age of 6 years old, with 1 converting after repeat SCT >60 mmol/L, 2 converting after the development and persistence of clinical symptoms, and 1 being re-classified after genetic mutation F191V classified as CF-causing. Patients 3 and 7 are siblings with the same genetic mutations, one of whom transitioned at 8 years old after their fifth SCT was positive at 61 mmol/L and repeat stool elastase found to be at 118, while the other sibling transitioned at 2 years old after developing clinical symptoms of cough, spirometry changes, upward trending SCT values, and oropharyngeal culture growth of methicillin-sensitive Staphylococcus aureus (MSSA) and Pseudomonas aeruginosa requiring eradication. Three patients had Pseudomonas aeruginosa growth on oropharyngeal cultures and 4 patients had MRSA growth on oropharyngeal cultures. None of the children with initial diagnosis of CRMS had evidence of hospitalizations for severe respiratory concerns prior to their transition to a CF diagnosis.
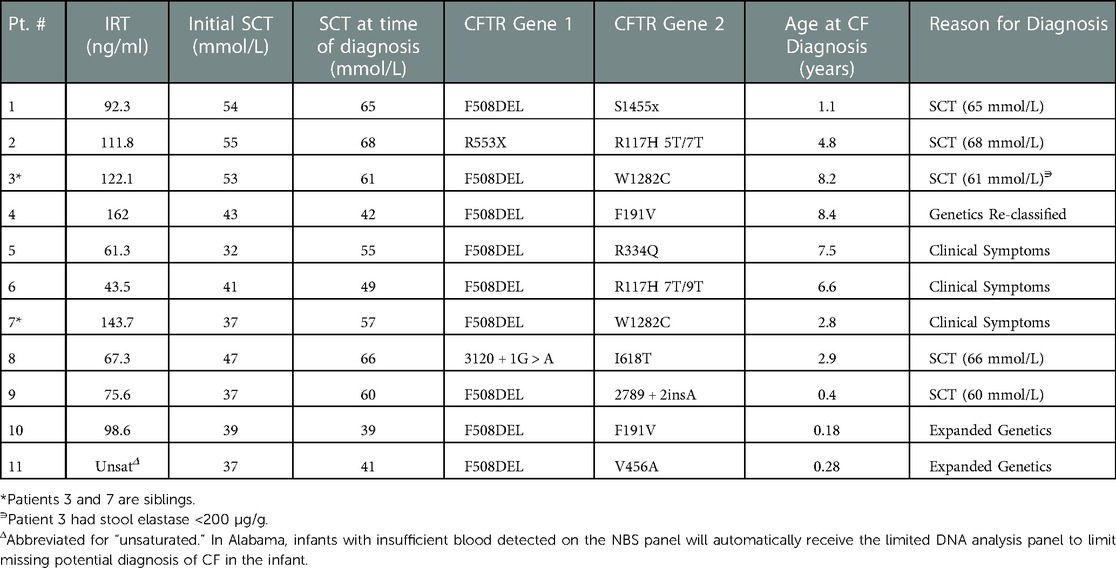
Table 3. Diagnostic data for infants transitioned to diagnosis of CF.
Biochemical biomarkers of cf diagnosis in CRMS sub-groups
In contrast to infants who were diagnosed with CF after completing the initial NBS process, infants initially diagnosed as CRMS and then transitioned later to a diagnosis of CF (CRMS-CF) had a higher, non-significant difference in the IRT value (97.8 ng/ml) on NBS compared to CRMS-P (75.2 ng/mL, p = 0.089) or CRMS-Carrier (67.4 ng/ml, p = 0.072) groups (Figure 2). The CRMS-CF group had a statistically significant elevation of their first SCT value (45 mmol/L) compared to CRMS-P (30 mmol/L, p < 0.0001) or CRMS-Carrier (35, p = 0.0279) groups. Because IRT was elevated in the CRMS-CF group (albeit non-significantly elevated), as was first SCT, we assessed inclusion of both IRT and SCT in the model to assess prediction of CF transition. First successful SCT was found to predict CRMS-P vs. CRMS-CF (p = 0.001) and CRMS-CF vs. CRMS-Carrier (p = 0.0149). Including IRT value does not further increase ability to predict CF transition. When we evaluated the trajectory of SCT repeated measures over time between the CRMS-CF and CRMS-P groups (the CRMS-Carrier having insufficient repeated measures to evaluate), we found that SCT concentrations increase with age (about 0.79 for every additional year of age, p = 0.014) over both groups. The mean first SCT concentration in the CRMS-CF group is higher than for CRMS-P and the CRMS-CF group increases more with age (1.23 mmol/L/year) compared to those in the CRMS-P group (0.49 mmol/L/year, p = 0.206). Stool elastase was collected from 38 children in the CRMS subgroups. Only one child had elastase <200 µg/g (transitioned to CRMS-CF) and otherwise no differences were appreciated between any of the subgroups.
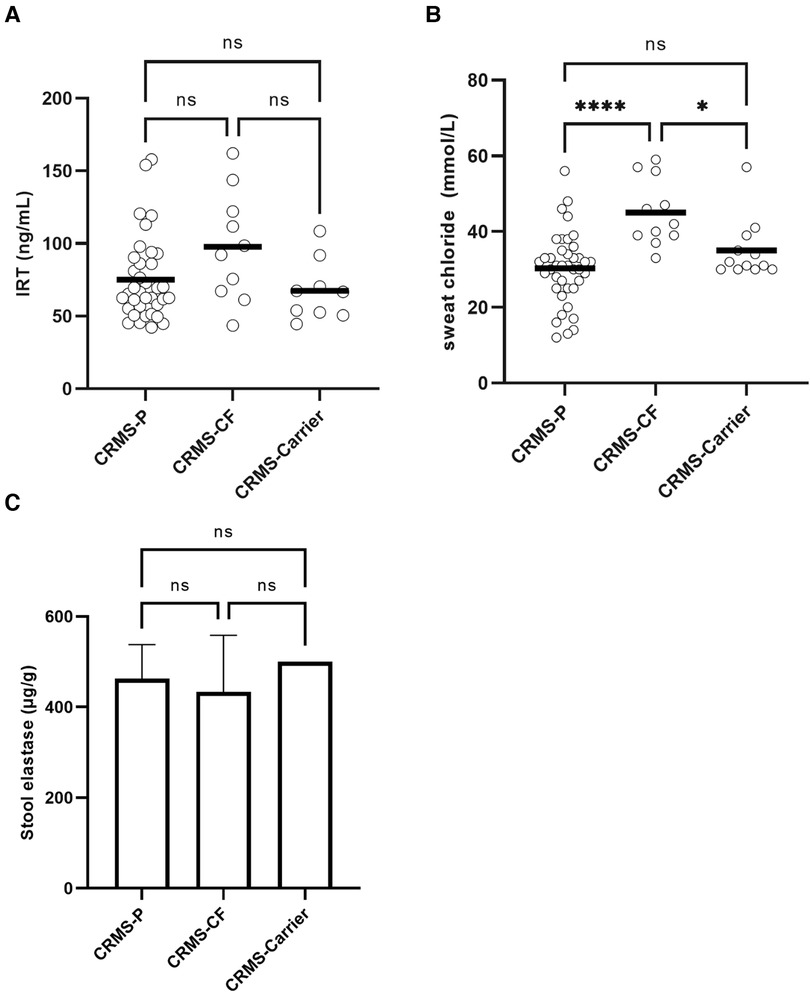
Figure 2. Biochemical biomarkers in CRMS sub-groups for IRT, initial SCT, and stool elastase. (A) Initial IRT results from NBS. A minority of patients had a result higher than the upper limit of quantitation, or an initial result that could not be determined. Comparisons are made between CRMS subgroups. (B) First successful SCT results by subgroup. First quantifiable results of SCT are shown. ****p < 0.0001, *p = 0.028. (C) Stool elastase results. The upper limit of quantitation of 500 µg/g stool is represented on the graph as 500 µg/g and was the result in 66% (25/38) available samples (including 6/10 CRMS-CF samples, 5/5 CRMS-Carrier samples, and 14/23 CRMS-P samples).
Microbiology of CRMS subgroups
A majority (76%) of children in the CRMS subgroups had a result for at least one sputum swab culture. Culture results were collected from all children with an initial designation of CRMS, with any cultures obtained after transition to a diagnosis of CF excluded. We had at least one culture in all of the children in the CRMS-CF group (range 1–16), 80% of children in the CRMS-P group (range 1–22), and 42% of children in the CRMS-Carrier group (range 1–2). There was a non-significant difference in the total number of cultures per patient collected between the CRMS-CF (mean = 5.5) vs. CRMS-P (mean = 3.4) groups. Results showing oropharyngeal flora and probable environmental contaminants were excluded, and number of unique pathogenic species ever cultured were normalized to number of cultures per child. Those in the CRMS-CF group had a slightly higher, non-significant number of unique species (0.75 species/culture) identified on microbiology compared to CRMS-Carrier (0.41 species/culture) and CRMS-P (0.61 species/culture) groups. Certain known CF-related pathogens (Pseudomonas aeruginosa, Methicillin-resistant Staphylococcus aureus, and Stenotrophomonas maltophilia) were isolated at least once in a greater proportion of children in the CRMS-CF group (18%, 45%, and 27%, respectively) compared to CRMS-P (5%, 25%, 7.5%) or CRMS-Carrier (8%, 17%, 0%) groups (Figure 3).
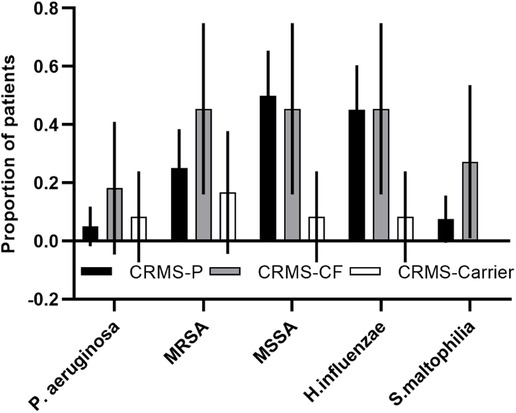
Figure 3. CF-related pathogens isolated in the CRMS subgroups. Proportion of children in each group that ever isolated the described bacteria are shown for each subgroup. 95% confidence intervals are shown for each.
Anthropomorphic measures
We examined weight-for-height, weight-for-age, and height-for-age using percentiles created by the CDC 2000 growth charts for children age 0 to 20 years old (26). These percentiles adjust for developmental differences among children in their anthropomorphic measurements by age and sex. Examining differences in these measures at first visit, we found no significant differences between CRMS-P, CRMS-CF, and CRMS-Carrier on weight-for-age-percentile or height-for-age-percentile, and no trend for CRMS-CF to be smaller than the other groups for these measures. When examining change over time in the anthropometric data between CRMS-CF and CRMS-P (there was insufficient data to evaluate CRMS-Carrier group), we found that children in the CRMS-P group have higher weight-for-height percentiles at first observation (59.7 [51.3,68.1] vs. 49.0 [24.0,74.0]) compared to children in the CRMS-CF group. Children in the CRMS-CF group appear to have a faster rate in growth than children in the CRMS-P group, but remain below the weight-for-height of those in CRMS-P by the time of the last observation (60.4 [43.5,77.3] vs. 67.0 [56.7,77.4]). Among children in the CRMS-CF group, each additional month is associated with an increase in weight-for-height percentile of 0.21 (p = 0.01), while children in the CRMS-P group have a non-significant change in weight-for-height percentile (−0.33, p = 0.695; difference between the two changes is 0.24, p=0.038). We do not see differences in their weight-for-age percentile or height-for-age percentile trajectories.
Discussion
Our single-center analysis identified that 18% (11 of the 63 total CRMS) of all children who met CRMS criteria on initial encounter transitioned to a conclusive diagnosis with CF, with 4 of these 11 children (36%) diagnosed after the age of 6 years of age. One-third of the children who transitioned were in the low intermediate range (30–39 mmol/L) on initial SCT, whereas the trajectory of SCT progression per year on repeated measurements was significantly greater in those who transitioned than those who did not. No child who transitioned to a diagnosis of CF had an initial SCT value in the normal range. No child who transitioned to CF demonstrated initial symptoms, and most were diagnosed while still asymptomatic on the basis of sweat test in the diagnostic range on repeat testing or genetics. While there were some modest differences in growth, there were no clear thresholds for predicting transition in diagnosis on the basis of growth parameters.
Biomarkers, such as IRT or SCT, may be helpful in predicting those who will transition to a CF diagnosis. Several studies have looked into the role initial and repeat SCT values may serve in predicting which infants with CRMS may potentially transition to a CF diagnosis (19, 20, 23). In our analysis, none of those with CRMS status with initial SCT values <30 mmol/L have so far transitioned to a conclusive CF diagnosis. Of the 11 children who transitioned from CRMS to a diagnosis of CF, all of them had an initial SCT value within the intermediate range. We found that 12% (4 out of 34 total) of children with CRMS status with initial SCT values between 30 and 39 mmol/L transitioned to CF. This is in contrast to 50% (4 out of  with initial SCT values between 40 and 49 mmol/L and 60% (3 out of 5 total) with initial SCT in the 50–59 mmol/L who ultimately received a diagnosis of CF.
with initial SCT values between 40 and 49 mmol/L and 60% (3 out of 5 total) with initial SCT in the 50–59 mmol/L who ultimately received a diagnosis of CF.
While IRT is useful for screening for CF, its utility alone to predict a transition to CF among those who initially meet CRMS criteria is low (27, 28). Despite this, our data shows that those who transitioned to a CF diagnosis had a higher IRT than the other subgroups, as well as a higher initial SCT. Furthermore, progressive increases in SCT over time may also be indicative of an eventual CF diagnosis, as has been reported previously (21, 24). Systematic evaluation of these biomarkers in multi-center studies is needed to define the risk and provide evidence for appropriate counseling and earlier diagnosis. With greater sample size, a predictive model using both IRT and initial SCT may prove useful for earlier diagnosis and should be evaluated.
Children with CRMS designation who transition to a CF diagnosis are not easily distinguishable from those who do not on the basis of clinical symptoms. Nine out of 11 of our CRMS-CF population remained pancreatic sufficient according to elastase results (and one unknown as the patient was followed elsewhere for care after diagnosis). There appears to be a modest difference in the isolation of pathogens, including CF-related pathogens before the CF diagnosis was confirmed in the CRMS-CF group. However, for most of these children, there were no concerns regarding chronic symptoms suggestive of CF prior to diagnosis. With a modestly higher isolation of CF-related pathogens identified in those who ultimately transitioned to a CF diagnosis, we speculate that appropriately powered, systematic studies of the colonization status in this population may provide greater insight into those who may need longer follow-up or more aggressive evaluation. There are also some modest differences in growth parameters. None of these present clear-cut thresholds to help with prognostic counseling.
There has been progress over the past ten years with respect to the evaluation, designation and early management of infants with CRMS (5, 23, 29). There is less clear evidence on long term management. The data from one study demonstrated that a significant proportion of asymptomatic children with CRMS reach 6 years of age in good health with normal growth, lung function and imaging and normal sweat chloride values (<30 mmol/L) and therefore are discharged from annual care at a CF Center (23). However, over one-third of children in our population transitioned to a CF diagnosis after the age of 6 years. Of these four children, two transitioned due to development of clinical symptoms and one due to repeat SCT in the diagnostic range. For populations similar to those in our study, continuing to monitor after age 6 years is warranted.
Despite the published and updated guidance, there is variation in practice and management for children with CRMS designation, possibly due to the lack of clear evidence supporting risk assessment and counseling. One of the unfortunate consequences of this inconsistent management is the potential for patients to be lost to follow-up. At our center, we identified 12 CRMS children that had been lost to follow-up between the years 2008 to 2019. In 2019, our institution implemented a center wide protocol based on updated guidelines, with our center re-engaging nearly 50% of that patient population for continued care. Given the data found in our analysis, we would emphasize the importance of consistency amongst CF providers at a center in ensuring this patient population is managed appropriately. Additionally, increased anxiety is exhibited by families for conditions associated with an inconclusive diagnosis, which can be ameliorated by providing clear information to families (30). While there is no internationally accepted consensus on the optimal approach to providing families with appropriate counseling for CRMS, there is evidence to support that CF Centers should incorporate counseling sessions with annual follow-ups to provide the family with awareness regarding any potential risk of transition to CF.
There are several limitations to our study. This study was conducted using retrospective chart review over a more than a decade, during which our institution underwent several changes in medical record keeping. Our analysis was conducted for a single state with one CF Center and therefore is associated with a small sample size. Over the 12 years of study evaluation, there were many different providers providing care, as well as changes in evidence-based guidelines for management. In our study, we retrospectively categorized each participant according to the updated 2017 diagnostic criteria to improve consistency across all years of evaluation. For many outcomes of interest, the practice variation resulted in missing data. For example, we were unable to identify one or more repeat SCT results for 8 (12.7%) of the patients, and other data such as culture results, fecal elastase, and growth parameters. Additionally, other states may use a different CF NBS algorithm or a different mutation screening panel, which may yield different results.
In conclusion, we describe a long-term, retrospective study of children designated CRMS and followed thereafter at an accredited CF Center. Our data complements previous studies that have noted a transition from CRMS designation to a conclusive CF diagnosis, while providing some important caveats to prior publications. Our results suggest that all infants with IRT+/DNA+ and a SCT within the intermediate range between 30 and 59 mmol/L need to be monitored closely for CF with strong consideration for repeat SCT, expanded genotyping, and microbiologic surveillance. Furthermore, children with elevated initial SCT, as well as those whose SCT is increasing in childhood, are likely higher risk for transition to a CF diagnosis. A multi-center effort to systematically study this population over childhood with harmonization of diagnostic criteria, evaluation, and follow-up should be initiated. This study should include assessment of current and novel biomarkers to better predict CFTR dysfunction as early as possible, especially with the advent of highly effective modulator therapy which may delay or prevent manifestations of CF disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary materials, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by UAB Institutional Review Board for Human Use. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author contributions
JSG: Conceptualization, Methodology, Formal Analysis, Investigation, Resources, Data Curation, Writing-Review and Editing, Visualization, Supervision, Project administration. MAG: Methodology, Formal Analysis, Investigation, Data Curation, Writing-Original Draft, Visualization. SS: Conceptualization, Investigation, Writing-Review and Editing. EB: Formal Analysis, Writing-Review and Editing. CM: Resources, Writing-Review and Editing. HG: Resources, Supervision, Funding Acquisition, Writing-Review and Editing. All authors read and commented on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MAG was supported by the U.S. Cystic Fibrosis Foundation (CFF) during her fellowship (#GUNNET19B0). JSG is supported by the NIH (K23HL143167) and the CFF (GUIMBE18A0-Q). JSG and EHB are supported by the CFF (GUIMBE22Y7). HG, SS, CM were supported by the CFF (CC032 to HG). HG and SS were supported by the CFF (GUTIER18QI0) and the Alabama Department of Public Health (GC-20-012/C00119005) for the Children’s of Alabama/University of Alabama at Birmingham Newborn Screening Program. All authors were supported by the Gregory Fleming James Cystic Fibrosis Center (R35HL135816, NIH DK072482, CFF UAB Research and Development Program—Rowe19RO) and the UAB Center for Clinical and Translational Science (UL1TR001417). Funding sources provided no input to the content of this manuscript.
Acknowledgments
We gratefully acknowledge the participants and families seen at our center actively for management of their cystic fibrosis disease and related conditions. We thank the COA CF Center’s pulmonary team for contributions to clinical care and data acquisition. We thank Kennedy Parker and Randi Gilinson for technical and administrative assistance.
Author disclaimer
This statement is to certify that all Authors have seen and approved the manuscript being submitted. We warrant that the article is the Authors’ original work. We warrant that the article has not received prior publication and is not under consideration for publication elsewhere. On behalf of all Co-Authors, the corresponding Author shall bear full responsibility for the submission. This research has not been submitted for publication nor has it been published in whole or in part elsewhere. We attest to the fact that all Authors listed on the title page have contributed significantly to the work, have read the manuscript, attest to the validity and legitimacy of the data and its interpretation. The work for this study was performed at the University of Alabama at Birmingham. None of the Authors have any conflicts of interest to disclose. No financial support was provided for development of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1127659/full#supplementary-material.
Abbreviations
CDC, centers for disease control and prevention; CF, cystic fibrosis; CFF, cystic fibrosis foundation; CFSPID, CF screen positive, inconclusive diagnosis; CFTR, cystic fibrosis transmembrane conductance regulator; COA, children’s of alabama; CRMS, CFTR-related metabolic syndrome; DNA, deoxyribonucleic acid; IRT, immunoreactive trypsinogen; MRSA, methicillin-resistant staphylococcus aeruginosa; MSSA, methicillin-sensitive staphylococcus aeruginosa; NBS, newborn screen; NTM, non-tuberculosis mycobacterium; SC, sweat chloride; SCT, sweat chloride test; UAB, university of alabama at birmingham; VCC, variable clinical consequence; SD, standard deviation; SEM, standard error of the mean.
References
3. Dijk FN, McKay K, Barzi F, Gaskin KJ, Fitzgerald DA. Improved survival in cystic fibrosis patients diagnosed by newborn screening compared to a historical cohort from the same centre. Arch Dis Child. (2011) 96(12):1118–23. doi: 10.1136/archdischild-2011-300449
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Farrell PM, White TB, Howenstine MS, Munck A, Parad RB, Rosenfeld M, et al. Diagnosis of cystic fibrosis in screened populations. J Pediatr. (2017) 181S:S33–S44.e2. doi: 10.1016/j.jpeds.2016.09.065
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Ren CL, Fink AK, Petren K, Borowitz DS, McColley SA, Sanders DB, et al. Outcomes of infants with indeterminate diagnosis detected by cystic fibrosis newborn screening. Pediatrics. (2015) 135(6):e1386–92. doi: 10.1542/peds.2014-3698
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation. J Pediatr. (2017) 181s:S4–S15.e1. doi: 10.1016/j.jpeds.2016.09.064
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Tosco A, Castaldo A, Colombo C, Claut L, Carnovale V, Iacotucci P, et al. Clinical outcomes of a large cohort of individuals with the F508del/5 T;TG12 CFTR genotype. J Cyst Fibros. (2022) 21(5):850–5. doi: 10.1016/j.jcf.2022.04.020
CrossRef Full Text | Google Scholar
9. Dolce D, Claut L, Colombo C, Tosco A, Castaldo A, Padoan R, et al. Different management approaches and outcome for infants with an inconclusive diagnosis following newborn screening for cystic fibrosis (CRMS/CFSPID) and Pseudomonas aeruginosa isolation. J Cyst Fibros. (2022) S1569-1993(22)00626-9. doi: 10.1016/j.jcf.2022.07.007
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Terlizzi V, Claut L, Tosco A, Colombo C, Raia V, Fabrizzi B, et al. A survey of the prevalence, management and outcome of infants with an inconclusive diagnosis following newborn bloodspot screening for cystic fibrosis (CRMS/CFSPID) in six Italian centres. J Cyst Fibros. (2021) 20(5):828–34. doi: 10.1016/j.jcf.2021.03.015
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Terlizzi V, Claut L, Colombo C, Tosco A, Castaldo A, Fabrizzi B, et al. Outcomes of early repeat sweat testing in infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/CF screen-positive, inconclusive diagnosis. Pediatr Pulmonol. (2021) 56(12):3785–91. doi: 10.1002/ppul.25683
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Sinha A, Southern KW. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). Breathe (Sheff). (2021) 17(3):210088. doi: 10.1183/20734735.0088-2021
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Barben J, Castellani C, Munck A, Davies JC, de Winter-de Groot KM, Gartner S, et al. Updated guidance on the management of children with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). J Cyst Fibros. (2021) 20(5):810–9. doi: 10.1016/j.jcf.2020.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Bauer SE, Wesson M, Oles SK, Ren CL. Outcomes of repeat sweat testing in cystic fibrosis newborn screen positive infants. Pediatr Pulmonol. (2021) 56(6):1521–6. doi: 10.1002/ppul.25296
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Gonska T, Keenan K, Au J, Dupuis A, Chilvers MA, Burgess C, et al. Outcomes of cystic fibrosis screening-positive infants with inconclusive diagnosis at school age. Pediatrics. (2021) 148(6):e2021051740. doi: 10.1542/peds.2021-051740
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Ooi CY, Castellani C, Keenan K, Avolio J, Volpi S, Boland M, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics. (2015) 135(6):e1377–85. doi: 10.1542/peds.2014-2081
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Levy H, Nugent M, Schneck K, Stachiw-Hietpas D, Laxova A, Lakser O, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. (2016) 89(5):539–49. doi: 10.1111/cge.12711
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kharrazi M, Yang J, Bishop T, Lessing S, Young S, Graham S, et al. Newborn screening for cystic fibrosis in California. Pediatrics. (2015) 136(6):1062–72. doi: 10.1542/peds.2015-0811
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Groves T, Robinson P, Wiley V, Fitzgerald DA. Long-term outcomes of children with intermediate sweat chloride values in infancy. J Pediatr. (2015) 166(6):1469–74.e1-3. doi: 10.1016/j.jpeds.2015.01.052
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Terlizzi V, Mergni G, Centrone C, Festini F, Taccetti G. Trend of sweat chloride values in a cohort of patients carrying CFTR mutations of varying clinical consequence: is there a risk of increasing sweat chloride over time? Pediatr Pulmonol. (2020) 55(5):1089–93. doi: 10.1002/ppul.24721
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Southern KW, Barben J, Gartner S, Munck A, Castellani C, Mayell SJ, et al. Inconclusive diagnosis after a positive newborn bloodspot screening result for cystic fibrosis; clarification of the harmonised international definition. J Cyst Fibros. (2019) 18(6):778–80. doi: 10.1016/j.jcf.2019.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Munck A, Bourmaud A, Bellon G, Picq P, Farrell PM. Phenotype of children with inconclusive cystic fibrosis diagnosis after newborn screening. Pediatr Pulmonol. (2020) 55(4):918–28. doi: 10.1002/ppul.24634
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Sosnay PR, Salinas DB, White TB, Ren CL, Farrell PM, Raraigh KS, et al. Applying cystic fibrosis transmembrane conductance regulator genetics and CFTR2 data to facilitate diagnoses. J Pediatr. (2017) 181:S27–S32.e1. doi: 10.1016/j.jpeds.2016.09.063
CrossRef Full Text | Google Scholar
27. Ooi CY, Sutherland R, Castellani C, Keenan K, Boland M, Reisman J, et al. Immunoreactive trypsinogen levels in newborn screened infants with an inconclusive diagnosis of cystic fibrosis. BMC Pediatr. (2019) 19(1):369. doi: 10.1186/s12887-019-1756-4
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Bombieri C, Seia M, Castellani C. Genotypes and phenotypes in cystic fibrosis and cystic fibrosis transmembrane regulator-related disorders. Semin Respir Crit Care Med. (2015) 36(2):180–93. doi: 10.1055/s-0035-1547318
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Borowitz D, Parad RB, Sharp JK, Sabadosa KA, Robinson KA, Rock MJ, et al. Cystic fibrosis foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J Pediatr. (2009) 155(6 Suppl):S106–16. doi: 10.1016/j.jpeds.2009.09.003
PubMed Abstract | CrossRef Full Text | Google Scholar
Актуальные вопросы диагностики муковисцидоза
Статьи
![]()
ЖУРНАЛ «ПРАКТИКА ПЕДИАТРА»
Опубликовано в журнале:
«ПРАКТИКА ПЕДИАТРА»; март-аперль; 2015; стр. 20-27.
Е.И. Кондратьева, д. м. н., профессор, В.Д. Шерман, к. м. н., Н.И. Капранов, д. м. н., профессор, Н.Ю. Каширская, д. м. н., профессор, НКО муковисцидоза ФГБНУ «МГНЦ», ГБУЗ «ДГКБ № 13 им. Н.Ф. Филатова ДЗМ», г. Москва
Муковисцидоз (МВ), или кистозный фиброз (cysticfibrosis), — одно из наиболее частых моногенных наследственных заболеваний с полиорганной патологией, резко сокращающее продолжительность и качество жизни пациентов без адекватного комплексного лечения в течение всей жизни. МВ распространен среди населения всей Земли, но наиболее часто поражает европеоидов: в среднем с частотой 1 на 2500-4500 новорожденных. Еще совсем недавно больные муковисцидозом умирали в раннем детском возрасте или даже на первом году жизни от пневмонии и истощения, обусловленными мальабсорбцией.
Ключевые слова: диагностика, генетика, мутации, неонатальный скрининг, потовая проба, эластаза кала.
Key words: cystic fibrosis, diagnosis, genetics, mutation, newborn screening, sweat test, fecal elastase.
Болезнь прежде всего характеризуется повышенной продукцией вязкого бронхиального секрета, частыми легочными инфекциями и обструкцией дыхательных путей. По мере прогрессирования легочной болезни образуются участки ателектазов, развивается эмфизема, постепенно разрушается паренхима легких с развитием бронхоэктазов и участков пневмосклероза, а больной имеет высокий риск погибнуть от легочно-сердечной недостаточности. В финальной стадии заболевания пересадка комплекса «сердце-легкие» остается для больного единственной надеждой. Помимо бронхолегочной системы у большинства больных муковисцидозом поражается поджелудочная железа, при этом это происходит внутриутробно. Недостаточность панкреатических ферментов обусловливает нарушение всасывания жиров и белков, развитие нутритивной недостаточности. В результате больные отстают в росте и страдают гипотрофией. Продукция инсулина также может быть нарушена, что ведет к развитию диабета. К частым осложнениям течения муковисцидоза относят остеопороз, а также жировой гепатоз с переходом в цирроз. При наличии «мягкой» мутации клинические проявления развиваются постепенно, преобладают моносимптомы, диагноз «муковисцидоз» устанавливается поздно или случайно.
Своевременная диагностика муковисцидоза, обеспечивающая в большинстве случаев раннее начало терапии, в том числе на доклиническом этапе, улучшает прогноз заболевания, повышает эффективность лечения, позволяет предупредить развитие тяжелых осложнений, значительного отставания в физическом развитии, а в ряде случаев и необратимых изменений в легких. Ранняя диагностика позволяет семье вовремя решить необходимые вопросы, связанные с рождением здорового ребенка (генетическое консультирование, пренатальная диагностика МВ в последующие беременности).
Диагностика делится на:
1) пренатальную диагностику;
2) диагностику по неонатальному скринингу (до клинических проявлений или при их дебюте);
3) диагностику при клинических проявлениях:
4) диагностику среди родственников больных.
В настоящее время налаживается дородовая диагностика муковисцидоза в перспективных и информативных семьях (Москва, Санкт-Петербург, Уфа, Томск, Красноярск, Ростов-на-Дону, Владивосток и некоторые другие города), что, безусловно, важно для профилактики этой тяжелой патологии. Пренатальная диагностика возможна в виде ДНК-диагностики при проведении амниоцентеза (получение околоплодных вод в ранний срок -13-14 недель и поздний — обычно 16-20 недель беременности) в семье носителей одной мутации гена CFTR и имеющей больного ребенка. Диагноз может быть заподозрен при УЗИ плода внутриутробно при наличии характерной УЗ-характеристики в виде гиперэхогенного кишечника. УЗИ во время беременности рекомендуют в скрининговые сроки: 11-14, 18-21 и 30-34 недели беременности. Обязательно проводят повторное исследование. В 50-78% случаев это состояние будет связано с МВ и проявится мекониальным илеусом. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высокоспецифичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями. При этом ДНК-диагностика родителей дает необходимую информацию о наличии мутаций у каждого из родителей и позволяет предполагать заболевание у ребенка при рождении.
Клинические признаки
1. Диагностика классической формы МВ обычно не представляет сложностей. Классический фенотип больного является результатом наличия двух мутантных копий гена муковисцидозного трансмембранного регулятора (CFTR) и характеризуется хронической бактериальной инфекцией дыхательных путей и придаточных пазух носа, стеатореей из-за внешнесекреторной недостаточности поджелудочной железы, мужским бесплодием из-за обструктивной азооспермии, а также повышенной концентрацией хлоридов потовой жидкости.
2. Проблемы диагностики МВ, как правило, связаны с фенотипическим разнообразием его форм, обусловленным генетическим полимор-
В ряде случаев атипичного течения МВ возможна его диагностика во взрослом возрасте. Как правило, в этой группе больных отмечается более мягкое течение болезни в связи с сохранностью функции поджелудочной железы и нетяжелым поражением органов дыхания.
В абсолютном большинстве случаев МВ может быть диагностирован в раннем детском возрасте (в 90% случаев — на первом году жизни). К сожалению, нередки случаи диагностики МВ у взрослых с классическим фенотипом.
Диагностика МВ у носителей «мягких» генотипов (актуально для детей, рожденных до 2006-2007 гг., и взрослых):
В настоящее время выделяют несколько групп риска по МВ.
Основной группой риска по заболеванию в РФ в настоящее время являются новорожденные с неонатальной гипертрипсиногенемией. Учитывая возможность получения ложноотрицательных результатов неонатального скрининга, а также то обстоятельство, что в РФ неонатальный скрининг на МВ проводится с 2006-2007 гг., не теряет своей актуальности анализ групп риска, включающих пациентов с патологией желудочно-кишечного тракта, бронхолегочными нарушениями, патологией других органов и родственников больных МВ (табл. 1).
Таблица 1.
Группы риска для дифференциальной диагностики муковисцидоза
| I. Бронхолегочные нарушения |
| 1. Повторные и рецидивирующие пневмонии с затяжным течением, особенно двусторонние 2. Бронхиальная астма, рефрактерная к традиционной терапии 3. Рецидивирующие бронхиты, бронхиолиты, особенно с высевом Ps. aeruginosa 4. Двусторонние бронхоэктазы |
| II. Изменения со стороны желудочно-кишечного тракта |
| 1. Синдром нарушенного кишечного всасывания неясного генеза 2. Мекониальный илеус и его эквиваленты 3. Гиперэхогенность кишечника плода 4. Желтуха обструктивного типа у новорожденных с затяжным течением 5. Цирроз печени 6. Сахарный диабет 7. Гастроэзофагеальный рефлюкс 8. Выпадение прямой кишки |
| III. Патология со стороны других органов |
| 1. Нарушение роста и развития 2. Задержка полового развития 3. Мужское бесплодие 4. Хронический синусит 5. Полипы носа 6. Электролитные нарушения |
| IV. Члены семей больных муковисцидозом |
Среди клинических проявлений, характерных для МВ, можно выделить высоко-и менее специфичные (табл. 2). Состояния, представленные в левой колонке таблицы, в абсолютном большинстве случаев встречаются у больных МВ. Причиной состояний из правой колонки могут быть другие заболевания, например первичная цилиарная дискинезия, гуморальный иммунодефицит и т. д.
Таблица 2.
Клинические проявления, характерные для МВ
| Высокоспецифичные для МВ | Менее специфичные для МВ |
| Желудочно-кишечные:
|
Желудочно-кишечные:
|
| Со стороны дыхательных путей:
|
Со стороны дыхательных путей:
|
| Другое:
|
Другое:
|
В таблице 3 представлены особенности проявлений МВ в разные возрастные периоды. Знание этих особенностей помогает специалистам, наблюдающим пациента с теми или иными симптомами, включить МВ в перечень заболеваний для дифференциальной диагностики. Особенно это касается детей раннего возраста, когда клиническая картина еще может быть неполной, но на себя будут обращать внимание некоторые проявления, например мекониальный илеус при рождении или синдром потери солей, не имеющий связи с патологией почек. Диагноз в этом случае может быть установлен еще до рождения ребенка. В то же время этот признак не является высоко специфичным для МВ, может быть транзиторным явлением, а также связанным с другими патологическими состояниями.
Таблица 3.
Клинические особенности проявлений МВ в различные возрастные периоды
| 0-2 года | |
|
|
|
| 3-16 лет | |
|
|
|
Диагностические критерии МВ
Для решения проблем диагностики МВ, в том числе и его атипичных форм, были разработаны критерии, согласно которым обязательным для МВ является наличие характерного клинического синдрома плюс доказательство какого-либо нарушения функции хлорного канала.
Учитывая все научные достижения в понимании природы муковисцидоза и МВ-зависимых заболеваний за последние 10 лет, в 2013 году группа экспертов Европейского общества муковисцидоза (European Cystic Fibrosis Society) под руководством Carlo Castellani подготовила новые стандарты диагностики в редакции Alan R. Smyth и Scott Bell (схема).
Схема.
Диагностические критерии муковисцидоза ECFS 2013
| Положительная потовая проба и/или две мутации МВТР, вызывающие МВ (согласно базе CFTR-2) |
И | Неонатальная гипертрипсиногенемия или характерные клинические проявления, такие как диффузные бронхоэктазы, высев из мокроты значимой для МВ патогенной микрофлоры (особенно синегнойной палочки), экзокринная панкреатическая недостаточность, синдром потери солей, обструктивная азооспермия |
Неонатальный скрининг
Проводится на основании Методических рекомендаций по проведению неонатального скрининга в РФ с использованием Европейских рекомендаций по неонатальному скринингу. 90% новорожденных без клинических проявлений муковисцидоза диагноз может быть установлен на основании скрининга в возрасте до 6 недель. В 5-10% случаев возникают трудности с диагностикой муковисцидоза (Cystic Fibrosis Foundation Patient Registry, 2005 Annual Data Report to the Center Directors. Bethesda, MD: CFF).
Проблемы неонатального скрининга:
Потовая проба
Показания:
1. При положительном результате неонатального скрининга (двукратном повышении уровня иммунореактивного трипсиногена в крови в течение первого месяца жизни ребенка).
2. При наличии у пациента каких-либо характерных клинических проявлений МВ.
3. Случаи МВ в семье.
Потовая проба является надежным методом диагностики МВ у 98% больных. Исследование можно проводить всем детям через 48 часов после рождения, хотя у новорожденных могут быть проблемы с набором пота. Несмотря на то, что «золотым стандартом» диагностики МВ считается количественное определение хлоридов в потовой жидкости (классический метод Гибсона — Кука), метод определения проводимости на аппаратах «Макродакт» и «Нанодакт» («Вескор», США) показал хорошую с ним корреляцию в многочисленных исследованиях.
Оценка результата
При положительном результате потовой пробы (хлориды > 60 ммоль/л при классическом методе Гибсона — Кука и/или проводимость > 80 ммоль/л NaCl) диагноз подтверждается.
Генетическое исследование
Генетическое исследование проводится после потовой пробы. Однако в связи с ограниченными возможностями ДНК-диагностики в России данный метод не является обязательным, однако применяется с исследовательской целью и для окончательного подтверждения диагноза.
На первом этапе ДНК-обследования наиболее часто используется панель, включающая 28 мутаций, как наиболее частых в мире, так и специфичных для России: F508del, CFTRdele2,3(21kb), 3849+10kbC>T, W1282X, 2143delT, 2184insA, 1677delTA, N1303K, G542X, R334W, E92K, L138ins, 394delTT, 3821delT, S1196X, 2789+5G>A, G85E, 2183AA>G, 604insA, 621+1G>T, R117H, R347P, R553X, 3667insTCAA, G551D, I507del, 1717-1G>A, 2184delA. По данным лаборатории генетической эпидемиологии ФГБУ «Медико-генетический научный центр» (МГНЦ) РАМН, при использовании данной панели удается обнаружить лишь около 82,5% мутантных аллелей у больных МВ. В случае когда при положительной потовой пробе не будет найдено ни одной мутации гена (что само по себе маловероятно), может потребоваться секвенирование гена МВ, позволяющее идентифицировать примерно 98% мутаций в гене CFTR.
Рекомендации:
1. На основании данных национального регистра больных МВ по ДНК-диагностике гена CFTR установлены особенности характера и частоты мутаций в регионах страны. На основе данных регистра рекомендуется создание региональных рекомендаций по определению мутаций со ссылкой на регистр (последнюю версию).
2. Отсутствие мутациий без проведения секвенирования — недостаточно для исключения МВ.
3. Некоторые мутации МВТР (3849+10 kb C>T) ассоциированы с нормальным или пограничным результатом потового теста.
4. «Мягкие» мутации характеризуются поздним дебютом заболевания, пограничным значением потовых проб, выявляются чаще при секвенировании.
5. Пациенты с пограничными результатами потовых проб (хлориды 30-60 ммоль/л и/или проводимость 50-80 ммоль/л), единственной мутацией гена представляют реальные трудности для диагностики.
Для диагностики МВ или его исключения при пограничных результатах пробы необходимо:
В европейских странах для подтверждения дефекта ионного транспорта применяется метод определения разности назальных потенциалов или измерение электрического тока в биоптате кишки, отражающие нарушение функции хлорного канала. Оба метода основаны на электрическом характере транспорта ионов и являются высокоинформативными для диагностики МВ.
Диагностика панкреатической недостаточности включает:
У больных МВ показатель эластазы может снижаться в течение первых лет жизни, поэтому определяется в динамике. Низкий уровень панкреатической эластазы расценивается как один из признаков МВ. Приблизительно 1% пациентов с МВ имеет пограничный результат потового теста в комплексе с сохранной функцией поджелудочной железы и хроническим бронхитом.
Диагностика хронического бронхолегочного процесса:
В качестве дополнительных диагностических маркеров могут быть использованы азооспермия в постпубертатном возрасте, идентификация МВ-ассоциированных патогенов из респираторного тракта, рентгенологические признаки синусита.
Знание основных симптомов МВ и особенностей его течения в разные возрастные периоды позволяет своевременно заподозрить наличие заболевания и направить пациента для дальнейшего обследования. Нередкие случаи поздней диагностики МВ связаны как с отсутствием у врачей достаточных знаний о заболевании, так и с фенотипическим разнообразием его форм. Ограниченные возможности ДНК-диагностики МВ в России и ее низкая доступность затрудняют и затягивают окончательную верификацию заболевания.
ЛИТЕРАТУРА
1. Муковисцидоз. Под ред. Н.И. Капранова, Н.Ю. Каширской. М.: ИД «МЕДПРАКТИКА-М», 2014, 672 с. ISBN 978-5-98803-314-1
2. Welsh M.J., Ramsey B.W., Accurso F.J., Cutting G.R. Cystic fibrosis. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill, 2001: 5121-88.
3. European cystic fibrosis society standards of care working group. Best practice guidelines. В редакции Alan R. Smith и Scott Bell, 2014.
4. Farell P.M., Rosenstein B.J., White T.B. et al. Cystic fibrosis foundation. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report // J. Pediatr., 2008; 153 (2): S4-S14.
5. Красовский С.А., Каширская Н.Ю., Усачева М.В., Амелина Е.Л., Черняк А.В., Науменко Ж.К. Влияние возраста постановки диагноза и начала специфической терапии на основные клинико-лабораторные проявления заболевания у больных муковисцидозом // Вопросы современной педиатрии, 2014, т. 13, № 2, с. 36-43.
6. de Boeck K., Wilschanski M., Castellani C. et al. Cystic fibrosis: terminology and diagnostic algorithms. Thorax, 2006; 61: 627-635.
7. de Oronzo M.A. Hyperechogenic fetal bowel: an ultrasonographic marker for adverse fetal and neonatal outcome? // J. Prenat. Med., 2011 Jan-Mar; 5 (1): 9-13.
8. Bombieri C. et al. Recommendations for the classification of diseases as CFTR-related disorders // Journal of Cystic Fibrosis, 2011, vol. 10, suppl. 2; S86-S102.
9. Hall E., Lapworth R. Use of sweat conductivity measurements. Annals of Clinical Biochemistry, 2010; 47: 390-392.
10. Sands D., Oltarzewski M., Nowakowska A., Zybert K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem Cystobiol., 2010 Sep 30; 48 (3): 358-65.
11. Sezer R.G., Aydemir G., Akcan A.B. et al. Nanoduct sweat conductivity measurements in 2664 patients: relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis // J. Clin. Med. Res., 2013 Feb; 5 (1): 34-41.
12. Петрова Н.В. Молекулярно-генетические и клинико-генотипические особенности муковисцидоза в российских популяциях. Автореф. дисс. докт. биол. наук. М., 2009, 42 с.
13. Derichs N., Sanz J., Von Kanel T. et al. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax, 2010 Jul; 65 (7): 594-9.
14. Servidoni M.F., Sousa M., Vinagre A.M. et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol., 2013 May 20; 13 (1): 91.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Муковисцидоз – наследственное врожденное заболевание, поражающее экзокринные железы и проявляющееся выработкой избытка слизистого секрета в организме. Из-за повышенного количества слизи страдают функции многих органов, и ребёнок с муковисцидозом нуждается в пожизненной терапии. Как выявляют муковисцидоз у детей? Рассказываем про симптомы, особенности диагностики и современные методы лечения.
Причины муковисцидоза
Патология развивается при наличии специфических мутаций в гене белка, ответственном за транспорт электролитов в протоках желез внешней секреции. Искажение генетической последовательности провоцирует возникновение структурных и функциональных нарушений синтеза белка, из-за чего слизь становится густой и плохо способной проходить через протоки. Возникает застой слизи, оптимальная среда для развития воспалительных процессов.
Муковисцидоз – заболевание наследственное, но родители могут не знать о носительстве мутации. Предупредить рецессивное наследование может консультация генетика, обследование до зачатия, а также репродуктивные технологии с тестированием эмбрионов при экстракорпоральном оплодотворении.
Формы муковисцидоза: какие органы страдают?

Выделяют несколько форм муковисцидоза в зависимости от выраженности поражения определенного органа: легочная форма, кишечная и смешанная. Кроме того, современная классификация учитывает также печеночную форму муковисцидоза, мекониальную непроходимость кишечника новорожденных, атипичный муковисцидоз – когда страдает только один орган, и стертую форму, симптомы которой могут долго оставаться нераспознанными.

Факт!
В четырех из пяти случаев развивается смешанная легочно-кишечная форма.
Симптомы у детей: на что обращать внимание
Муковисцидоз у детей разного возраста проявляется различными симптомокомплексами.
Период новорожденности и младенчества
Чаще всего болезнь проявляет себя наличием сильного кашля (нередко до рвоты), одышкой, удушьем. Муковисцидоз в менее выраженной форме может проявляться длительным восстановлением массы тела, недостатком набора веса.
Еще один симптом, характерный для муковисцидоза у новорожденных, – закупорка кишечника меконием. Мекониальный илеус, непроходимость первородного кала, встречается в одном случае из пяти при раннем начале заболевания.
Важно!
Без срочного оказания помощи мекониальный илеус приводит к жизнеугрожающим состояниям: завороту кишок или прободению кишечника.
В возрасте старше 10 суток от рождения частыми признаками муковисцидоза у детей становятся атония (вялость), отказ от сосания, избыток газов, длительная желтуха новорожденных, рвота желчью. Кожа сухая, бледная, недостаточно упругая, на вкус соленая (этот признак лег в основу старейшего лабораторного теста на муковисцидоз).
Муковисцидоз у грудных детей
По информации экспертов Союза педиатров России, муковисцидоз чаще всего выявляют у детей в возрасте до двух лет. В последние годы благодаря введению скрининга на это заболевание в обязательный список анализов в родильном доме (скрининг «пяточка») возраст постановки диагноза снизился.
Факт!
Чаще всего первые симптомы у грудных детей проявляются в возрасте полугода после введения прикорма или перевода ребёнка с грудного молока на смесь.
Симптомы муковисцидоза могут быть похожи на длительные и частые респираторные или кишечные инфекции, регулярные нарушения пищеварения. У грудных детей болезнь проявляется следующими признаками:
- Появление густого кала с жирным блеском и неприятным запахом;
- Задержки физического, а затем психоэмоционального развития;
- Одышка, кашель, приступы рвоты;
- Сухая кожа с сероватым оттенком.
В раннем детстве муковисцидоз у детей может провоцировать частые бронхиты или пневмонии с переходом в хронический бронхолегочный воспалительный процесс, из-за чего грудная клетка деформируется, становится килевидной, воронкообразной, бочкообразной формы.
Кишечная форма проявляется менее ярко в первые месяцы, а порой и годы жизни: болезнь вызывает ферментную недостаточность из-за закупорки протоков поджелудочной железы, что приводит к смазанной симптоматике, под которую подпадают многие диагнозы.
Факт!
В России муковисцидоз диагностируется у одного из 9 000 новорожденных детей.
Дошкольный и старший школьный возраст: проявления муковисцидоза

В данных возрастных группах чаще всего наблюдаются следующие симптомы муковисцидоза:
- Частые приступы кашля с выводом гнойной мокроты;
- Одышка;
- Разжижение каловых масс;
- Случаи обезвоживания;
- Выпадение прямой кишки, непроходимость кишечника;
- Изменение формы фаланг пальцев на руках («барабанные палочки»);
- Хронические синуситы и т. д.
Насторожить должны также панкреатит и признаки страдания печени у ребёнка, развитие сахарного диабета.
Муковисцидоз в подростковом периоде
В период пубертата к перечисленным выше проявлениям заболевания добавляется задержка полового развития, патологическое снижение выносливости при физических нагрузках. Поражение печени может привести к циррозу, асциту, расширению вен пищевода, часты гастриты, язвы желудка, воспалительные процессы в пищеводе и учащенная дефекация.
Диагностика муковисцидоза в России
- Первый этап диагностики в последние годы проводится в родильном отделении в рамках скрининга новорожденных, обязательной медицинской процедуры. Болезнь может подтвердиться на разных этапах скрининга.
- Первый этап – забор капли крови из пяточки ребёнка на 4-7 сутки после рождения.
- Второй этап (21-28 сутки после рождения) проводят при повышенном количестве иммунореактивного трипсина в высушенном пятне крови.
- Если на втором этапе результаты скрининга опять положительные, требуется потовая проба и генетический анализ, так как скрининг на муковисцидоз может при определенных условиях давать ложноположительный результат.
Третий этап позволяет полностью подтвердить или опровергнуть диагноз. В диагностический план также могут включаться рентгенография грудной клетки, бронхоскопия, бронхография, копрограмма, анализ мокроты и т. д.
Лечение муковисцидоза: современные методы терапии

Муковисцидоз на данный момент – пожизненное неизлечимое заболевание, требующее постоянной и комплексной терапии для облегчения состояния и профилактики осложнений.
Базовая терапия включает медикаментозные препараты – антибиотики, муколитики, ферментные препараты, витамины; регулярные упражнения дыхательной гимнастики, диету, специальные аппаратные методы, облегчающие вывод слизи из легких и бронхов и так далее. Тяжелое поражение легкого или печени требует трансплантации.
Комплекс мер, направленных на вывод мокроты, назначается пожизненно и проводится ежедневно. В него входят аэрозольные ингаляции, лечебная гимнастика, вибрационный массаж, постуральный дренаж. Упражнения и постуральный дренаж показаны не менее одного раза в день, вибрационный массаж проводят от трех раз в сутки.
Из спектра муколитических препаратов отдают предпочтение дорназе альфа, гипертоническому раствору натрия хлорида, ингаляционному маннитолу и некоторым другим препаратам.
Дорназа альфа – базовый муколитический препарат в терапии муковисцидоза. Ингаляции с препаратом назначают сразу же после положительного результата диагностики.
Дорназа альфа – фермент, аналог дезоксирибонуклеазы I (ДНКазы I), в норме присутствующей в организме каждого человека в достаточном количестве. Генно-инженерный вариант ДНКазы I, дорназа альфа, применяется в симптоматической терапии заболевания почти 30 лет, и по оценке специалистов считается наиболее эффективным и безопасным средством для облегчения состояния и профилактики инфекционно-воспалительных процессов, возникающих при застое секрета.
Применение дорназы альфа вызывает разрушение внеклеточной ДНК, а значит, эффективно разжижает скопившуюся мокроту, облегчая ее вывод. Препарат достоверно снижает смертность пациентов на 15% и является незаменимой частью терапии муковисцидоза. До его разработки у врачей и пациентов не было достаточно надежного средства для эффективной муколитической терапии.
Важно!
Все больные муковисцидозом в РФ имеют право на обеспечение дорназой альфа за счет государства.
Федеральные клинические рекомендации по оказанию медицинской помощи детям с кистозным фиброзом (муковисцидозом) / Баранов А.А., Намазова-Баранова Л.С., Симонова О.И. и др. – 2020
Муковисцидоз у детей / Ивкина С.С., Кривицкая Л.В., Латохо Т.А. // Проблемы здоровья и экологии. – 2015 – №4 (46).
Эффективность и безопасность биоаналогичного лекарственного препарата Тигераза® (дорназа альфа) при длительной симптоматической терапии пациентов с муковисцидозом: результаты клинического исследования III фазы / Е. Л. Амелина, С. А. Красовский, Д. И. Абдулганиева, и др. // Пульмонология – 2019 – Том 29, № 6

Прочтите и возьмите себе на заметку, особенно если вы молодые люди
В России уже много лет проводится массовое обследование новорожденных для выявления у них нескольких наследственных заболеваний. Такое обследование проводится во многих странах и называется скринингом новорожденных или неонаталъным скринингом.
Целью скрининга новорожденных является, конечно, не само выявление новорожденных с еще не проявившимися наследственными заболеваниями, а их лечение, которое позволяет предотвратить появление клинических симптомов, во многих случаях весьма тяжелых, или даже фатальных. В результате рано начатого и аккуратно проводимого лечения вместо тяжело больных детей, а затем подростков и взрослых, получаются здоровые люди, полноценные члены общества, нередко являющиеся гордостью семьи.
Скрининг новорожденных в России ведется в отношении 5 наследственных и врожденных заболеваний: фенилкетонурии, гипотиреоза, галактоземии, адрено-гениталъного синдрома и муковисцидоза.
ЧТО ТАКОЕ МУКОВИСЦИДОЗ?
Муковисцидоз — это наследственное заболевание, обусловленное изменением (мутацией) в гене, который отвечает за синтез белка, осуществляющего в клетках функцию канала для ионов хлора. Из-за нарушения функции этого канала слизь и другие секреты становятся очень густыми и вязкими в легких, поджелудочной железе и других органах. Это приводит к развитию в легких хронической инфекции, повреждающей легочную ткань; нарушению переваривания пищи, поскольку ферменты поджелудочной железы не могут попасть в кишечник, и другим клиническим проявлениям.
КАК НАСЛЕДУЕТСЯ МУКОВИСЦИДОЗ?
Муковисцидоз наследуется по аутосомно-рецессивному типу, когда больные в семье появляются только в одном поколении. Схема такого наследования приведена на рисунке, на котором изображен фрагмент родословной семьи, в которой родился ребенок, больной муковисцидозом. На родословной мужчины обозначены квадратиком, а женщины — кружочком. Внутри этих квадратиков и кружочков нарисована только одна хромосома (из 23 пар, имеющихся у человека), несущая нормальный или мутантный ген муковисцидоза, который помечен черной точкой.
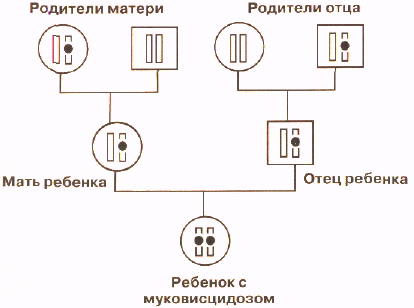
На рисунке для простоты изображена только хромосома, содержащая ген, мутации в котором вызывают муковисцидоз. У ребенка в обеих хромосомах содержится мутантный ген и поэтому он болен. У каждого из родителей мутантный ген содержится только в одной хромосоме, а вторая хромосома нормальная и поэтому они здоровы. Такие люди, которые имеют один нормальный и один дефектный ген называются носителями мутантного гена. У бабки по матери мутантный ген также имеется только в одной хромосоме, как и у деда со стороны отца. Они, как и родители ребенка, здоровы, но передали хромосомы, содержащие мутантный ген, своим детям. У вторых деда и бабки обе хромосомы содержат только нормальный ген. Таким образом, при рецессивном наследовании болен только тот член семьи, который получил от своих родителей обе хромосомы, несущие мутантный ген. Все остальные члены семьи здоровы, в том числе и те, кто является носителем мутантного гена. На схеме родословной видно, что у родителей больного ребенка могут еще появиться больные дети. Вероятность появления больного ребенка в семьях, в которых родители являются носителями мутантного гена, составляет 1/4 или 25%. Эта вероятность не меняется от числа больных или здоровых детей в семье: для каждого следующего ребенка риск, что он будет болен, составляет 25%. Вероятность рождения здорового ребенка, обе хромосомы которого содержат только нормальный ген, составляет также 25%. А 50% детей будут иметь один нормальный и один мутангный ген, как их родители. Это означает, что при каждой беременности родители-носители имеют шансы 3 из 4 (75%) родить здорового ребенка. Многие родители больных муковисцидозом детей и их родственники, первый раз встретившись с врачом-генетиком, настойчиво повторяют, что у их ребенка не наследственное заболевание, так как в их семье ни у кого из родственников никогда не было такого заболевания. Только объяснение, что правила наследования бывают разные, и не редко больной с наследственным заболеванием может быть единственным в семье, позволяют им понять с какой ситуацией они столкнулись.
КАКИЕ ПОРАЖЕНИЯ В ОРГАНИЗМЕ ВЫЗЫВАЕТ МУКОВИСЦИДОЗ?
Заболевание обычно начинается в раннем возрасте. Клиницисты различают три основные формы муковисцидоза: легочную, кишечную и смешанную. Самой частой из них является смешанная форма. Она встречается примерно у 80% больных муковисцидозом. Легочная форма муковисцидоза проявляется хроническим обструктивным бронхолегочным процессом. Из-за того, что мокрота у больных густая и вязкая, она не отхаркивается. Большое количество белка в мокроте делает ее хорошей средой для развития разных микробов, в том числе стафилококков и синегнойной палочки. Развивается хронический воспалительный процесс, приводящий к разрушению легочной ткани. Кровь больных плохо насыщается кислородом, из-за чего начинают страдать сердце, печень и другие . органы. Лечение больных с легочной формой муковисцидоза требует применения мощных антибиотиков в больших дозах. При кишечной форме муковисцидоза нарушается процесс переваривания пищи, так как ферменты поджелудочной железы, расщепляющие белки и жиры, не попадают в кишечник вследствие закупорки протоков железы. Больные отстают от своих родственников в росте и весе. Основное лечение кишечной формы заключается в приеме ферментов поджелудочной железы. Эффективность этого лечения легко контролируется по количеству жира в кале ребенка. Препараты поджелудочной железы должны даваться в таком количестве, чтобы жира в кале не было. При смешанной форме муковисцидоза кишечные проявления муковисцидоза усугубляют поражение легких. Лечение смешанной формы наиболее сложное. У больных муковисцидозом, не получающих необходимого лечения, продолжительность жизни, как правило, относительно короткая.
СУЩЕСТВУЮТ ЛИ ТЕСТЫ НА РАННЕЕ ВЫЯВЛЕНИЕ МУКОВИСЦИДОЗА?
Больные с муковисцидозом могут быть выявлены с помощью скрининга новорожденных. В отличие от других наследственных болезней, выявляемых при скрининге, при которых раннее выявление заболевания позволяет его настолько эффективно лечить, что можно вообще избежать каких-либо клинических проявлений заболеваний, скрининг новорожденных при муковисцидозе преследует несколько иную цель. Врачи считают, что если муковисцидоз выявляется у новорожденного, и его начинают сразу же лечить, то ребенок нормально развивается физически и умственно. У него в меньшей степени будут поражаться легкие и другие органы, увеличится продолжительность жизни, которая в настоящее время, благодаря адекватному лечению, составляет в развитых странах более 35 лет. Скрининг на муковисцидоз начинается с того, что у новорожденного в родильном доме перед выпиской берут из пятки несколько капель крови, которую наносят на специально для этой цели используемую фильтровальную бумагу. Кровь высушивается, и такой бланк, на котором указана фамилия новорожденного и ряд других сведений, необходимых для его идентификации, переправляется в лабораторию региональной медико-генетической консультации. В лаборатории проводят специальное исследование, которое позволяет выявить новорожденных, у которых есть подозрение на муковисцидоз. В этом случае ребенок через педиатра вызывается в лабораторию на повторноетестирование. Родителям педиатр сообщает, что первый тест на муковисцидоз у их ребенка оказался ненормальным. У них появляется повод для беспокойства. Поэтому повторное тестирование образца крови у младенца, которое является очень важным, нужно сделать как можно быстрее. В большинстве случаев при повторном исследовании тест на муковисцидоз оказывается нормальным. Это означает, что результат первого исследования был неверный (его называют ложноположительным). Причины этого могут быть разными и связанными как с состоянием младенца, так и с какой-то ошибкой лаборатории. Этот результат, свидетельствующий о том, что у ребенка нет муковисцидоза, сразу же сообщается родителям, чтобы снять с них чувство страха.
ЧТО ДЕЛАТЬ, ЕСЛИ И ПОВТОРНЫЙ ЛАБОРАТОРНЫЙ ТЕСТ НА МУКОВИСЦИДОЗ ОКАЗАЛСЯ ПОЛОЖИТЕЛЬНЫМ?
Если и второе лабораторное исследование для выявления муковисцидоза оказалось положительным, то, в отличие от других скринируемых наследственных болезней, это еще не означает, что у ребенка есть муковисцидоз, хотя вероятность такого диагноза является высокой. Семья, в которой у ребенка повторно подтвердился положительный тест на муковисцидоз, приглашается на прием к врачу-генетику в медико-генетическую консультацию. Здесь семье объясняют, что собой представляет муковисцидоз и организуют прием у клиницистов, являющихся специалистами в диагностике и лечении муковисцидоза. Теперь наблюдение за ребенком берет на себя группа клиницистов. Для подтверждения диагноза муковисцидоза младенцу проводят так называемый лотовый тест. Это совершенно безобидный и безболезненный тест можно провести младенцу, начиная с возраста 3-4 недели жизни. Если лотовый тест оказывается отрицательным, то ребенок считается здоровым, хотя за ним клиницисты еще будут наблюдать некоторое время. Если же потовый тест оказался положительным, то диагноз муковисцидоза считается установленным, даже до появления каких-либо клинических проявлений заболевания. В этом случае врачи назначат ребенку лечение, которое надо будет строго соблюдать. Ребенок будет периодически обследоваться группой специалистов для постоянного контроля за состоянием его здоровья.
МОЖНО ЛИ ПОМОЧЬ СЕМЬЕ, В КОТОРОЙ ПОЯВИЛСЯ БОЛЬНОЙ С МУКОВИСЦИДОЗОМ, ИМЕТЬ ЗДОРОВЫХ ДЕТЕЙ?
Да, и довольно успешно. Для муковисцидоза возможна дородовая диагностика. Первым шагом в этом направлении является обращение в медико-генетическую консультацию, где врач-генетик определяет показания и возможные методические подходы к дородовой диагностике в каждом конкретном случае. Сама процедура заключается в том, что во время беременности в сроке 9-11 недель или 16-18 недель врач акушер-гинеколог проводит забор очень небольшого количества клеток плода и направляет этот материал в специальную лабораторию пренатальной диагностики. В этой лаборатории врачи лаборанты-генетики проводят молекулярную диагностику, т.е. определяют наличие или отсутствие мутации в гене, отвечающем за муковисцидоз. В случае положительного результата семья решает вопрос о прерывании беременности больным плодом или настраивается на появление еще одного больного ребенка. Это право выбора остается за семьей.