| Treacher Collins syndrome | |
|---|---|
| Other names | Treacher Collins–Franceschetti syndrome,[1] mandibulofacial dysostosis,[2] Franceschetti-Zwalen-Klein syndrome[3] |
 |
|
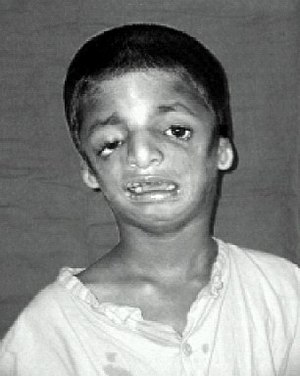
| Child with Treacher Collins syndrome[4] | |
| Specialty | Medical genetics |
| Symptoms | Deformities of the ears, eyes, cheekbones, chin[5] |
| Complications | Breathing problems, problems seeing, hearing loss[5] |
| Causes | Genetic[5] |
| Diagnostic method | Based on symptoms, X-rays, genetic testing[3] |
| Differential diagnosis | Nager syndrome, Miller syndrome, hemifacial microsomia[3] |
| Treatment | Reconstructive surgery, hearing aids, speech therapy[6] |
| Prognosis | Generally normal life expectancy[6] |
| Frequency | 1 in 50,000 people[5] |
Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin.[5] The degree to which a person is affected, however, may vary from mild to severe.[5] Complications may include breathing problems, problems seeing, cleft palate, and hearing loss.[5] Those affected generally have normal intelligence.[5]
TCS is usually autosomal dominant.[5] More than half the time it occurs as a result of a new mutation rather than being inherited.[5] The involved genes may include TCOF1, POLR1C, or POLR1D.[5] Diagnosis is generally suspected based on symptoms and X-rays, and potentially confirmation by genetic testing.[3]
Treacher Collins syndrome is not curable.[6] Symptoms may be managed with reconstructive surgery, hearing aids, speech therapy, and other assistive devices.[6] Life expectancy is generally normal.[6] TCS occurs in about one in 50,000 people.[5] The syndrome is named after Edward Treacher Collins, an English surgeon and ophthalmologist, who described its essential traits in 1900.[7][8]
Signs and symptoms[edit]

Symptoms in people with Treacher Collins syndrome vary. Some individuals are so mildly affected that they remain undiagnosed, while others have moderate to severe facial involvement and life-threatening airway compromise.[9] Most of the features of TCS are symmetrical and are already recognizable at birth.[citation needed]
The most common symptom of Treacher Collins syndrome is underdevelopment of the lower jaw and underdevelopment of the zygomatic bone. This can be accompanied by the tongue being retracted. The small mandible can result in a poor occlusion of the teeth or in more severe cases, trouble breathing or swallowing. The respiratory system of a child with Treacher Collins syndrome is the primary concern at birth, with other issues only addressed once respiratory function has been stabilized.[10] Underdevelopment of the zygomatic bone gives the cheeks a sunken appearance.[11][12]
The external ear is sometimes small, rotated, malformed, or absent entirely in people with TCS. Symmetric, bilateral narrowing or absence of the external ear canal, is also described.[12][13] In most cases, the bones of the middle ear and the middle ear cavity are misshapen. Inner ear malformations are rarely described. As a result of these abnormalities, a majority of the individuals with TCS have conductive hearing loss.[12][14]
Most affected people also experience eye problems, including coloboma (notches) in the lower eyelids, partial or complete absence of eyelashes on the lower lid, downward angled eyelids, drooping of upper and lower eyelids, and narrowing of the tear ducts. Vision loss can occur and is associated with strabismus, refractive errors, and anisometropia. It can also be caused by severely dry eyes, a consequence of lower eyelid abnormalities and frequent eye infections.[12][13][15][16]
Although an abnormally shaped skull is not distinctive for Treacher Collins syndrome, brachycephaly with bitemporal narrowing is sometimes observed.[13] Cleft palate is also common.[12]
Dental anomalies are seen in 60% of affected people, including tooth agenesis (33%), discoloration (enamel opacities) (20%), malplacement of the maxillary first molars (13%), and wide spacing of the teeth. In some cases, dental anomalies in combination with mandible hypoplasia result in a malocclusion. This can lead to problems with food intake and the ability to close the mouth.[12]
Less common features of TCS may add to an affected person’s breathing problems, including sleep apnea. Choanal atresia or stenosis is a narrowing or absence of the choanae, the internal opening of the nasal passages, which may also be observed. Underdevelopment of the pharynx can also narrow the airway.[12]
Features related to TCS that are seen less frequently include nasal deformities, high-arched palate, macrostomia, preauricular hair displacement, cleft palate, hypertelorism, notched upper eyelid, and congenital heart defects.[11][12][16]
Although facial deformity is often associated with developmental delay and intellectual disability, more than 95% of people affected with TCS have normal intelligence.[12] The psychological and social problems associated with facial deformity can affect quality of life in individuals with TCS.
Genetics[edit]

Mutations in TCOF1, POLR1C, or POLR1D genes can cause Treacher Collins syndrome.[17] TCOF1 gene mutations are the most common cause of the disorder, with POLR1C and POLR1D gene mutations causing an additional 2% of cases. In individuals without an identified mutation in one of these genes, the genetic cause of the condition is unknown. The TCOF1, POLR1C, and POLR1D genes code for proteins which play important roles in the early development of bones and other tissues of the face. Mutations in these genes reduce the production of rRNA, which may trigger the self-destruction (apoptosis) of certain cells involved in the development of facial bones and tissues. It is unclear why the effects of a reduction in rRNA are limited to facial development. Mutations in TCOF1 and POLR1D cause the autosomal dominant form of Treacher Collins, and mutations in POLR1C cause the autosomal recessive form.[12]
TCOF1[edit]
TCOF1 is the primary gene associated with TCS, a mutation in this gene being found in 90–95% of the individuals with TCS.[11][18] However, in some individuals with typical symptoms of TCS, mutations in TCOF1 have not been found.[19] Investigation of the DNA has resulted in the identification of the kind of mutations found in TCOF1. The majority of mutations are small deletions or insertions, though splice site and missense mutations also have been identified.[11][20][21][22]
Mutation analysis has unveiled more than 100 disease-causing mutations in TCOF1, which are mostly family-specific mutations. The only recurrent mutation accounts for about 17% of the cases.[23]
TCOF1 is found on the 5th chromosome in the 5q32 region. It codes for a relatively simple nucleolar protein called treacle, that is thought to be involved in ribosome assembly.[18] Mutations in TCOF1 lead to haploinsufficiency of the treacle protein.[24] Haploinsufficiency occurs when a diploid organism has only one functional copy of a gene, because the other copy is inactivated by a mutation. The one normal copy of the gene does not produce enough protein, causing disease. Haploinsufficiency of the treacle protein leads to a depletion of the neural crest cell precursor, which leads to a reduced number of crest cells migrating to the first and second pharyngeal arches. These cells play an important role in the development of the craniofacial appearance, and loss of one copy of treacle affects the cells’ ability to form the bones and tissues of the face.[12][20][25]
Other mutations[edit]
POLR1C and POLR1D mutations are responsible for a minority of cases of Treacher Collins. POLR1C is found on chromosome 6 at position 6q21.2 and POLR1D is found on chromosome 13 at position 13q12.2. Those genes code for a protein subunits shared between RNA polymerase I and III. Both of these polymerases are important for ribosome biogenesis.[12]
Diagnosis[edit]
Genetic counseling[edit]
TCS is inherited in an autosomal dominant manner and the penetrance of the affected gene is almost complete.[26] Some recent investigations, though, described some rare cases in which the penetrance in TCS was not complete. Causes may be a variable expressivity, an incomplete penetrance[27] or germline mosaicism.[28] Only 40% of the mutations are inherited. The remaining 60% are a result of a de novo mutation, where a child has a new mutation in the responsible gene and did not inherit it from either parent.[12][29] In the outcome of the disease, inter- and intrafamilial variability occurs. This suggests that when an affected child is born, it is important to investigate the parents to determine whether the affected gene is present, because the parent could have a mild form of the disease that has not been diagnosed. In this case, the risk of having another affected child is 50%. If the parents do not have the affected gene, the recurrence risk appears to be low.[26] In following generations, the severity of the clinical symptoms increases.[21]
Prenatal diagnosis[edit]
Mutations in the main genes responsible for TCS can be detected with chorionic villus sampling or amniocentesis. Rare mutations may not be detected by these methods. Ultrasonography can be used to detect craniofacial abnormalities later in pregnancy, but may not detect milder cases.[12]
Clinical findings[edit]
TCS is often first suspected with characteristic symptoms observed during a physical exam. However, the clinical presentation of TCS can resemble other diseases, making diagnosis difficult.[30] The OMENS classification was developed as a comprehensive and stage-based approach to differentiate the diseases. This acronym describes five distinct dysmorphic manifestations, namely orbital asymmetry, mandibular hypoplasia, auricular deformity, nerve development, and soft-tissue disease.[31]
Orbital symmetry
- O0: normal orbital size, position
- O1: abnormal orbital size
- O2: abnormal orbital position
- O3: abnormal orbital size and position
Mandible
- M0: normal mandible
- M1: small mandible and glenoid fossa with short ramus
- M2: ramus short and abnormally shaped
- 2A: glenoid fossa in anatomical acceptable position
- 2B: Temperomandibular joint inferiorly (TMJ), medially, anteriorly displaced, with severely hypoplastic condyle
- M3: Complete absence of ramus, glenoid fossa, and TMJ
Ear
- E0: normal ear
- E1: Minor hypoplasia and cupping with all structures present
- E2: Absence of external auditory canal with variable hypoplasia of the auricle
- E3: Malposition of the lobule with absent auricle, lobular remnant usually inferior anteriorly displaced
Facial nerve
- N0: No facial nerve involvement
- N1: Upper facial nerve involvement (temporal or zygomatic branches)
- N2: Lower facial nerve involvement (buccal, mandibular or cervical)
- N3: All branches affected
Soft tissue
- S0: No soft tissue or muscle deficiency
- S1: Minimal tissue or muscle deficiency
- S2: Moderate tissue or muscle deficiency
- S3: Severe tissue or muscle deficiency
Radiographs[edit]
A few techniques are used to confirm the diagnosis in TCS.[30][32]
An orthopantomogram (OPG) is a panoramic dental X-ray of the upper and lower jaw. It shows a two-dimensional image from ear to ear. Particularly, OPG facilitates an accurate postoperative follow-up and monitoring of bone growth under a mono- or double-distractor treatment. Thereby, some TCS features could be seen on OPG, but better techniques are used to include the whole spectrum of TCS abnormalities instead of showing only the jaw abnormalities.[30]
Another method of radiographic evaluation is taking an X-ray image of the whole head. The lateral cephalometric radiograph in TCS shows hypoplasia of the facial bones, like the malar bone, mandible, and the mastoid.[30]
Finally, occipitomental radiographs are used to detect hypoplasia or discontinuity of the zygomatic arch.[32]
CT scan[edit]
A temporal-bone CT using thin slices makes it possible to diagnose the degree of stenosis and atresia of the external auditory canal, the status of the middle ear cavity, the absent or dysplastic and rudimentary ossicles, or inner ear abnormalities such as a deficient cochlea. Two- and three-dimensional CT reconstructions with VRT and bone and skin-surfacing are helpful for more accurate staging and the three-dimensional planning of mandibular and external ear reconstructive surgery.[citation needed]
Differential diagnosis[edit]
Other diseases have similar characteristics to Treacher Collins syndrome. In the differential diagnosis, one should consider the acrofacial dysostoses. The facial appearance resembles that of Treacher Collins syndrome, but additional limb abnormalities occur in those persons. Examples of these diseases are Nager syndrome and Miller syndrome.[citation needed]
The oculoauriculovertebral spectrum should also be considered in the differential diagnosis. An example is hemifacial microsomia, which primarily affects development of the ear, mouth, and mandible. This anomaly may occur bilaterally. Another disease which belongs to this spectrum is Goldenhar syndrome, which includes vertebral abnormalities, epibulbar dermoids and facial deformities.[33]
Treatment[edit]
The treatment of individuals with TCS may involve the intervention of professionals from multiple disciplines. The primary concerns are breathing and feeding, as a consequence of the hypoplasia of the mandibula and the obstruction of the hypopharynx by the tongue. Sometimes, they may require a tracheostomy to maintain an adequate airway,[34] and a gastrostomy to assure an adequate caloric intake while protecting the airway. Corrective surgery of the face is performed at defined ages, depending on the developmental state.[35]
An overview of the present guidelines:
- If a cleft palate is present, the repair normally takes place at 9–12 months old. Before surgery, a polysomnography with a palatal plate in place is needed. This may predict the postoperative situation and gives insight on the chance of the presence of sleep apnea (OSAS) after the operation.[11][36][37]
- Hearing loss is treated by bone conduction amplification, speech therapy, and educational intervention to avoid language/speech problems. The bone-anchored hearing aid is an alternative for individuals with ear anomalies.[38]
- Zygomatic and orbital reconstruction is performed when the cranio-orbitozygomatic bone is completely developed, usually at the age of 5–7 years. In children, an autologous bone graft is mostly used. In combination with this transplantation, lipofilling can be used in the periorbital area to get an optimal result of the reconstruction.[citation needed] Reconstruction of the lower eyelid coloboma includes the use of a myocutaneous flap, which is elevated and in this manner closes the eyelid defect.[39]
- External ear reconstruction is usually done when the individual is at least eight years old. Sometimes, the external auditory canal or middle ear can also be treated.
- The optimal age for the maxillomandibular reconstruction is controversial; as of 2004, this classification has been used:[11]
- Type I (mild) and Type IIa (moderate) 13–16 years
- Type IIb (moderate to severe malformation) at skeletal maturity
- Type III (severe) 6–10 years
- When the teeth are cutting, the teeth should be under supervision of an orthodontist to make sure no abnormalities occur. If abnormalities like dislocation or an overgrowth of teeth are seen, appropriate action can be undertaken as soon as possible.[20]
- Orthognathic treatments usually take place after the age of 16 years; at this point, all teeth are in place and the jaw and dentition are mature. Whenever OSAS is detected, the level of obstruction is determined through endoscopy of the upper airways. Mandibular advancement can be an effective way to improve both breathing and æsthetics, while a genioplasty only restores the profile.[11]
- If a nose reconstruction is necessary, it is usually performed after the orthognathic surgery and after the age of 18 years.[11]
- The contour of the facial soft tissues generally requires correction at a later age, because of the facial skeletal maturity. The use of microsurgical methods, like the free flap transfer, has improved the correction of facial soft tissue contours.[40] Another technique to improve the facial soft tissue contours is lipofilling. For instance, lipofilling is used to reconstruct the eyelids.[39]
Hearing loss[edit]
Hearing loss in Treacher Collins syndrome is caused by deformed structures in the outer and middle ear. The hearing loss is generally bilateral with a conductive loss of about 50–70 dB. Even in cases with normal auricles and open external auditory canals, the ossicular chain is often malformed.[41]
Attempts to surgically reconstruct the external auditory canal and improve hearing in children with TCS have not yielded positive results.[42]
Auditory rehabilitation with bone-anchored hearing aids (BAHAs) or a conventional bone conduction aid has proven preferable to surgical reconstruction.[38]
Psychiatric[edit]
The disorder can be associated with a number of psychological symptoms, including anxiety, depression, social phobia, and distress about body image. People who have this disorder may also experience discrimination, bullying, and name calling, especially when young. A multi-disciplinary team and parental support should include these issues.[43]
Epidemiology[edit]
TCS occurs in about one in 50,000 births in Europe.[44] Worldwide, it is estimated to occur in one in 10,000 to one in 50,000 births.[12]
History[edit]
The syndrome is named after Edward Treacher Collins (1862–1932), the English surgeon and ophthalmologist who described its essential traits in 1900.[7][8][45] In 1949, Adolphe Franceschetti and David Klein described the same condition on their own observations as mandibulofacial dysostosis. The term mandibulofacial dysostosis is used to describe the clinical features.[46]
Culture[edit]
In July 1977, a New York Times article describing new plastic surgery techniques which could partially correct the appearance of those with Treacher Collins syndrome was widely circulated resulting in raised awareness of the disease.[47]
Prior to beginning his comedy career, Bob Saget made a documentary short called «Through Adam’s Eyes» documenting his young nephew’s experiences undergoing facial reconstructive surgery due to Treacher Collins; the film won a student Academy Award.[48]
The disorder was featured on the show Nip/Tuck, in the episode «Blu Mondae».[49] TLC’s Born Without a Face[50] features Juliana Wetmore, who was born with the most severe case in medical history of this syndrome and is missing 30%–40% of the bones in her face.[50]
In 2010, BBC Three documentary Love Me, Love My Face[51] covered the case of a man, Jono Lancaster, with the condition. In 2011, BBC Three returned to Jono to cover his and his partner Laura’s quest to start a family,[2] in So What If My Baby Is Born Like Me?,[52] which first aired as part of a BBC Three season of programmes on parenting.[53] The first film was replayed on BBC One shortly ahead of the second film’s initial BBC Three broadcast. Lancaster’s third BBC Three film, Finding My Family on Facebook, which looked at adoption, aired in 2011.[54]
In Wonder, a children’s novel, the main character is a child who has Treacher Collins syndrome.[55] A 2017 film adaptation of Wonder, starring Julia Roberts, Owen Wilson and Jacob Tremblay, was released in November 2017.[56][57]
Alison Midstokke, who appears in the drama film Happy Face (2018),[58] is an actress and activist who has the condition.
See also[edit]
- First arch syndrome
- Franceschetti-Klein syndrome
- Hearing loss with craniofacial syndromes
References[edit]
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 894, 1686. ISBN 978-1-4160-2999-1.
- ^ a b «I hated seeing my face in the mirror». BBC Online. 18 November 2010. Archived from the original on 2018-10-11. Retrieved 2010-11-18.
- ^ a b c d «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). 2016. Archived from the original on 20 December 2019. Retrieved 7 November 2017.
- ^ a b Goel, L; Bennur, SK; Jambhale, S (August 2009). «Treacher-collins syndrome-a challenge for anaesthesiologists». Indian Journal of Anaesthesia. 53 (4): 496–500. PMC 2894488. PMID 20640217.
- ^ a b c d e f g h i j k l «Treacher Collins syndrome». Genetics Home Reference. June 2012. Archived from the original on 26 June 2020. Retrieved 7 November 2017.
- ^ a b c d e «Treacher Collins syndrome». rarediseases.info.nih.gov. 2015. Archived from the original on 16 August 2019. Retrieved 7 November 2017.
- ^ a b R, Pramod John; John, Pramod (2014). Textbook of Oral Medicine. JP Medical Ltd. p. 76. ISBN 9789350908501. Archived from the original on 2020-09-12. Retrieved 2017-09-17.
- ^ a b Beighton, Greta (2012). The Man Behind the Syndrome. Springer Science & Business Media. p. 173. ISBN 9781447114154.
- ^ Edwards, S J; Fowlie, A; Cust, M P; Liu, D T; Young, I D; Dixon, M J (1 July 1996). «Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging». Journal of Medical Genetics. 33 (7): 603–606. doi:10.1136/jmg.33.7.603. PMC 1050672. PMID 8818950.
- ^ Mouthon, L., Busa, T., Bretelle, F., Karmous‐Benailly, H., Missirian, C., Philip, N., & Sigaudy, S. (2019). Prenatal diagnosis of micrognathia in 41 fetuses: Retrospective analysis of outcome and genetic etiologies. American Journal of Medical Genetics Part A, 179(12), 2365–2373. doi: 10.1002/ajmg.a.61359
- ^ a b c d e f g h Katsanis SH, et al., Treacher Collins syndrome, 2004, GeneReviews
- ^ a b c d e f g h i j k l m n o «The Physician’s Guide to Treacher Collins Syndrome» (PDF). National Organization for Rare Disorders (NORD). 2012. Archived from the original (PDF) on 2017-01-28.
- ^ a b c Posnick, Jeffrey C (1 October 1997). «Treacher Collins syndrome: Perspectives in evaluation and treatment». Journal of Oral and Maxillofacial Surgery. 55 (10): 1120–1133. doi:10.1016/S0278-2391(97)90294-9. PMID 9331237.
- ^ Trainor, Paul A; Dixon, Jill; Dixon, Michael J (24 December 2008). «Treacher Collins syndrome: etiology, pathogenesis and prevention». European Journal of Human Genetics. 17 (3): 275–283. doi:10.1038/ejhg.2008.221. PMC 2986179. PMID 19107148.
- ^ Hertle, R W; Ziylan, S; Katowitz, J A (1 October 1993). «Ophthalmic features and visual prognosis in the Treacher-Collins syndrome». British Journal of Ophthalmology. 77 (10): 642–645. doi:10.1136/bjo.77.10.642. PMC 504607. PMID 8218033.
- ^ a b Marszałek, B; Wójcicki, P; Kobus, K; Trzeciak, WH (2002). «Clinical features, treatment and genetic background of Treacher Collins syndrome». Journal of Applied Genetics. 43 (2): 223–33. PMID 12080178.
- ^ «Treacher Collins Syndrome». NORD (National Organization for Rare Disorders). Archived from the original on 2019-12-20. Retrieved 2016-02-29.
- ^ a b Dixon, Jill; Edwards, Sara J.; Gladwin, Amanda J.; Dixon, Michael J.; Loftus, Stacie K.; Bonner, Cynthia A.; Koprivnikar, Kathryn; Wasmuth, John J. (31 January 1996). «Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome». Nature Genetics. 12 (2): 130–136. doi:10.1038/ng0296-130. PMID 8563749. S2CID 34312227.
- ^ Teber OA, Gillessen-Kaesbach G, Fischer S, Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, König R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D (2004). «Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation». European Journal of Human Genetics. 12 (11): 879–90. doi:10.1038/sj.ejhg.5201260. PMID 15340364.
- ^ a b c Dixon, J; Trainor, P.; Dixon, M. J. (1 May 2007). «Treacher Collins syndrome». Orthodontics & Craniofacial Research. 10 (2): 88–95. doi:10.1111/j.1601-6343.2007.00388.x. PMID 17552945.
- ^ a b Masotti, Cibele; Ornelas, Camila C.; Splendore-Gordonos, Alessandra; Moura, Ricardo; Félix, Têmis M.; Alonso, Nivaldo; Camargo, Anamaria A.; Passos-Bueno, Maria (1 January 2009). «Reduced transcription of TCOF1 in adult cells of Treacher Collins syndrome patients». BMC Medical Genetics. 10 (1): 136. doi:10.1186/1471-2350-10-136. PMC 2801500. PMID 20003452.
- ^ Sakai, Daisuke; Trainor, Paul A. (31 May 2009). «Treacher Collins syndrome: Unmasking the role of Tcof1/treacle». The International Journal of Biochemistry & Cell Biology. 41 (6): 1229–1232. doi:10.1016/j.biocel.2008.10.026. PMC 3093759. PMID 19027870.
- ^ Splendore, Alessandra; Fanganiello, Roberto D.; Masotti, Cibele; Morganti, Lucas S. C.; Rita Passos-Bueno, M. (1 May 2005). «TCOF1 mutation database: Novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature». Human Mutation. 25 (5): 429–434. doi:10.1002/humu.20159. PMID 15832313. S2CID 12500736.
- ^ Isaac, C.; Marsh, K. L.; Paznekas, W. A.; Dixon, J.; Dixon, M. J.; Jabs, E. W.; Meier, U. T. (September 2000). «Characterization of the nucleolar gene product, treacle, in Treacher Collins syndrome». Molecular Biology of the Cell. 11 (9): 3061–3071. doi:10.1091/mbc.11.9.3061. PMC 14975. PMID 10982400.
- ^ Gorlin RJ, Syndromes of the Head and Neck, 2001, Oxford University Press, 4th edition
- ^ a b Dixon, MJ; Marres, HA; Edwards, SJ; Dixon, J; Cremers, CW (April 1994). «Treacher Collins syndrome: correlation between clinical and genetic linkage studies». Clinical Dysmorphology. 3 (2): 96–103. doi:10.1097/00019605-199404000-00002. PMID 8055143.
- ^ Dixon, Jill; Ellis, Ian; Bottani, Armand; Temple, Karen; Dixon, Michael James (15 June 2004). «Identification of mutations in TCOF1: Use of molecular analysis in the pre- and postnatal diagnosis of Treacher Collins syndrome». American Journal of Medical Genetics. 127A (3): 244–248. doi:10.1002/ajmg.a.30010. PMID 15150774. S2CID 2091796.
- ^ Shoo, Brenda A.; McPherson, Elizabeth; Jabs, Ethylin Wang (1 April 2004). «Mosaicism of aTCOF1 mutation in an individual clinically unaffected with treacher collins syndrome». American Journal of Medical Genetics. 126A (1): 84–88. doi:10.1002/ajmg.a.20488. PMID 15039977. S2CID 35163245.
- ^ Splendore, Alessandra; Jabs, Ethylin Wang; Félix, Têmis Maria; Passos-Bueno, Maria Rita (31 August 2003). «Parental origin of mutations in sporadic cases of Treacher Collins syndrome». European Journal of Human Genetics. 11 (9): 718–722. doi:10.1038/sj.ejhg.5201029. PMID 12939661.
- ^ a b c d Senggen, E; Laswed, T; Meuwly, JY; Maestre, LA; Jaques, B; Meuli, R; Gudinchet, F (May 2011). «First and second branchial arch syndromes: multimodality approach» (PDF). Pediatric Radiology. 41 (5): 549–61. doi:10.1007/s00247-010-1831-3. PMID 20924574. S2CID 22416094. Archived (PDF) from the original on 2019-04-26. Retrieved 2020-01-15.
- ^ Vento AR, et al., The O.M.E.N.S classification of hemifacial microsomia, 1991, Cleft Palate Craniofac, J 28, p. 68-76
- ^ a b Posnick JC; et al. (2000). «Treacher Collins syndrome: current evaluation, treatment, and future directions». Cleft Palate Craniofac J. 55 (5): 1120–1133. doi:10.1597/1545-1569(2000)037<0434:TCSCET>2.0.CO;2. PMID 11034023.
- ^ Dixon, MJ (1995). «Treacher Collins syndrome». Journal of Medical Genetics. 32 (10): 806–8. doi:10.1136/jmg.32.10.806. PMC 1051706. PMID 8558560.
- ^ Goel L; et al. (2009). «Treacher Collins syndrome-a challenge for anaesthesiologists». Indian J Anaesth. 53 (4): 642–645. PMC 2894488. PMID 20640217.
- ^ Evans, Adele Karen; Rahbar, Reza; Rogers, Gary F.; Mulliken, John B.; Volk, Mark S. (31 May 2006). «Robin sequence: A retrospective review of 115 patients». International Journal of Pediatric Otorhinolaryngology. 70 (6): 973–980. doi:10.1016/j.ijporl.2005.10.016. PMID 16443284.
- ^ Rose, Edmund; Staats, Richard; Thissen, Ulrike; Otten, Jörg-Eland; Schmelzeisen, Rainer; Jonas, Irmtrud (1 August 2002). «Sleep-Related Obstructive Disordered Breathing in Cleft Palate Patients after Palatoplasty». Plastic and Reconstructive Surgery. 110 (2): 392–396. doi:10.1097/00006534-200208000-00002. PMID 12142649. S2CID 36499038.
- ^ Bannink, Natalja; Mathijssen, Irene M. J.; Joosten, Koen F. M. (1 September 2010). «Use of Ambulatory Polysomnography in Children With Syndromic Craniosynostosis». Journal of Craniofacial Surgery. 21 (5): 1365–1368. doi:10.1097/SCS.0b013e3181ec69a5. PMID 20856022. S2CID 43739792.
- ^ a b Marres, HA (2002). Hearing loss in the Treacher-Collins syndrome. pp. 209–15. doi:10.1159/000066811. ISBN 978-3-8055-7449-5. PMID 12408086.
- ^ a b Zhang, Zhiyong; Niu, Feng; Tang, Xiaojun; Yu, Bing; Liu, Jianfeng; Gui, Lai (1 September 2009). «Staged Reconstruction for Adult Complete Treacher Collins Syndrome». Journal of Craniofacial Surgery. 20 (5): 1433–1438. doi:10.1097/SCS.0b013e3181af21f9. PMID 19816274. S2CID 44847925.
- ^ Saadeh, Pierre B.; Chang, Christopher C.; Warren, Stephen M.; Reavey, Patrick; McCarthy, Joseph G.; Siebert, John W. (1 June 2008). «Microsurgical Correction of Facial Contour Deformities in Patients with Craniofacial Malformations: A 15-Year Experience». Plastic and Reconstructive Surgery. 121 (6): 368e–378e. doi:10.1097/PRS.0b013e3181707194. PMID 18520863. S2CID 27971712.
- ^ Argenta, Louis C.; Iacobucci, John J. (30 June 1989). «Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction». World Journal of Surgery. 13 (4): 401–409. doi:10.1007/BF01660753. PMID 2773500. S2CID 27094477.
- ^ Marres, HA; Cremers, CW; Marres, EH (1995). «Treacher-Collins syndrome. Management of major and minor anomalies of the ear». Revue de Laryngologie – Otologie – Rhinologie. 116 (2): 105–108. PMID 7569369.
- ^ Jane Ridley (Aug 12, 2022). «Man’s parents rejected him at birth because of his appearance». Insider. Retrieved 2022-08-13.
- ^ Conte, Chiara; Maria Rosaria D’Apice; Fabrizio Rinaldi; Stefano Gambardella; Federica Sanguiuolo; Giuseppe Novelli (27 September 2011). «Novel mutations of TCOF1 gene in European patients with treacher Collins syndrome». Medical Genetics. 12: 125. doi:10.1186/1471-2350-12-125. PMC 3199234. PMID 21951868.
- ^ Treacher Collin E (1900). «Cases with symmetrical congenital notches in the outer part of each lid and defective development of the malar bones». Trans Ophthalmol Soc UK. 20: 190–192.
- ^ Franceschetti A, Klein D (1949). «Mandibulo-facial dysostosis: new hereditary syndrome». Acta Ophthalmol. 27: 143–224.
- ^ Jr, Donald G. McNeil (1977-07-26). «Surgical Teamwork Gives Disease Victims a New Life». The New York Times. ISSN 0362-4331. Retrieved 2023-01-25.
- ^ «Bob Saget Biography». Retrieved 2022-06-30.
{{cite web}}: CS1 maint: url-status (link) - ^ «Nip/Tuck: Blu Mondae — TV.com». Archived from the original on 2008-06-12. Retrieved 2008-05-12.
- ^ a b «First Coast News: Local Family Has Daughter Born Without a Face».
- ^ «BBC programme page for Love Me, Love My Face». BBC Three. 17 June 2011. Archived from the original on 21 January 2018. Retrieved 7 November 2017.
- ^ «BBC programme page for So What If My Baby…» BBC Three. 24 August 2011. Archived from the original on 2 June 2017. Retrieved 7 November 2017.
- ^ «BBC Three Bringing Up Britain season». BBC One. 12 April 2011. Archived from the original on 12 August 2011. Retrieved 24 August 2011.
- ^ «Finding My Family on Facebook». bbc.co.uk. BBC Three. 2011. Archived from the original on 2011-10-31..
- ^ Chilton, Martin (24 February 2012). «Wonder by R. J. Palacio: review». The Telegraph. Archived from the original on 6 October 2017. Retrieved 7 November 2017.
- ^ «Julia Roberts’ Drama ‘Wonder’ Pushed to November». The Hollywood Reporter. February 13, 2017. Archived from the original on April 22, 2017. Retrieved February 13, 2017.
- ^ O’Conner, Katie (December 22, 2017). «Real Life Reflected on the Silver Screen». Richmond Times-Dispatch.
- ^ Wilner, Norman (February 18, 2020). «Canadian Screen Awards 2020: Prepare for a Schitt’s show». Now. Archived from the original on 2020-02-18. Retrieved December 28, 2020.
External links[edit]
Синдром Тричера-Коллинза (Мандибулофациальный дизостоз, Синдром Тричера Коллинза-Франческетти, Челюстно-лицевой дизостоз)

Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.

Синдром Тричера-Коллинза
Причины
Развитие синдрома в 78-93% случаев обусловлено мутациями гена TCOF1, расположенного на пятой хромосоме в регионе 5q32. Данный ген кодирует производство ядерного фосфопротеина Treacle. У 7-9% пациентов причиной заболевания является дефект гена POLR1C, локализованного на шестой хромосоме, или гена POLR1D, находящегося на тринадцатой хромосоме. Они ответственны за синтез I и III РНК-полимеразы.
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
- Осмотр, сбор анамнеза. Определяются характерные черепно-лицевые аномалии: недоразвитость костей скул и челюсти, деформация и гипоплазия ушных раковин, антимонголоидный тип глазных щелей, нарушения слуха и дефект верхнего неба. Иногда подтвержденный диагноз синдрома имеется у одного из родителей.
- Биогенетический тест. Антенатальное исследование включает молекулярный анализ образца ворсин хориона на 10-11 неделе беременности, фетоскопию и анализ крови из сосудов плаценты на 18-20 неделе. После родов выполняется забор крови из вены ребенка. В обоих случаях исследуется ген TCOF1. Заболевание подтверждается при наличии в нем мутации любого типа.
- Дородовое УЗИ. С 20-24 недели беременности ультразвуковое исследование плода способно выявить типичные изменения лица. Наиболее четко заметна двусторонняя аномалия ушей, гипоплазия скул и челюсти.
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
|
Литература 1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009. 2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 — №3. |
Код МКБ-10 Q75.4 |
Синдром Тричера-Коллинза — лечение в Москве
Синдром Тричера-Коллинза
Содержимое
- 1 Синдром Тричера-Коллинза
- 1.1 Что такое синдром Тричера-Коллинза
- 1.2 Основные симптомы синдрома Тричера-Коллинза
- 1.3 Влияние на психическое состояние человека
- 1.4 Причины развития синдрома Тричера-Коллинза
- 1.5 Генетическая предрасположенность к синдрому Тричера-Коллинза
- 1.6 Внутренние и внешние факторы, способствующие развитию синдрома
- 1.7 Диагностика синдрома Тричера-Коллинза
- 1.8 Профилактика и предотвращение синдрома Тричера-Коллинза
- 1.9 Методы лечения синдрома Тричера-Коллинза
- 1.10 Видео по теме:
- 1.11 Вопрос-ответ:
-
- 1.11.0.1 Что такое синдром Тричера-Коллинза?
- 1.11.0.2 Какие симптомы характерны для синдрома Тричера-Коллинза?
-
- 1.12 Последствия нераспознанного и нелеченного синдрома Тричера-Коллинза
- 1.13 Современные исследования и перспективы лечения синдрома Тричера-Коллинза
Синдром Тричера-Коллинза — редкое генетическое заболевание, которое характеризуется различными физическими и психическими особенностями. Узнайте о причинах, симптомах и методах лечения этого синдрома.
Синдром Тричера-Коллинза, также известный как синдром Дюпюи-Генеза, является редким генетическим заболеванием, которое влияет на развитие лица и конечностей у новорожденных. Этот синдром был впервые описан в 1963 году французскими врачами А. Тричером и П. Коллинзом. Главными характеристиками этого синдрома являются аномалии лица, такие как необычная форма носа, расщелина верхней губы и галечкообразные уши. Также у детей, страдающих от этого синдрома, наблюдаются деформации конечностей, проблемы с пищеварением и кардиоваскулярные аномалии.
Синдром Тричера-Коллинза является генетическим нарушением, вызванным делецией, или удалением, определенной части хромосомы 8. Несмотря на то, что причина этой делеции неизвестна, исследования показали, что она происходит случайно, без наличия наследственности или других известных факторов риска. Это означает, что риск развития синдрома не возрастает у детей, у которых родители уже имеют этот синдром.
На данный момент не существует специфического лечения для синдрома Тричера-Коллинза. Однако доступны различные методы поддерживающего лечения, которые могут помочь улучшить качество жизни пациентов с этим синдромом. Хирургические вмешательства могут быть необходимы для коррекции аномалий лица и конечностей. Также рекомендуется проводить регулярные медицинские обследования, чтобы контролировать состояние сердца и других органов.
Что такое синдром Тричера-Коллинза

Синдром Тричера-Коллинза, также известный как синдром Тричера-Коллинза или синдром Тричера-Коллинза, является генетическим расстройством, которое проявляется в различных физических и психических особенностях.
Основным признаком синдрома Тричера-Коллинза является ретардация интеллекта, то есть задержка умственного развития. Это означает, что люди с этим синдромом имеют низкий уровень когнитивных способностей и обычно нуждаются в постоянной поддержке и уходе.
Кроме того, синдром Тричера-Коллинза характеризуется особыми физическими особенностями, такими как низкий рост, короткая шея и широкий диапазон лицевых дефектов. У пациентов может быть также задержка развития речи, проблемы с видением и слухом, а также другие медицинские проблемы, включая сердечные и желудочно-кишечные заболевания.
Основные симптомы синдрома Тричера-Коллинза
1. Задержка психомоторного развития: дети с синдромом Тричера-Коллинза обычно достигают моторных и психических мильников позже, чем их сверстники. Способности к ходьбе, говорению и когнитивному развитию могут быть отставать на несколько лет.
2. Физическое нарушение роста: дети с этим синдромом могут иметь низкий рост и низкий вес по сравнению со сверстниками. Также, у них может быть маленькая голова, маленький размер лица и особые черты лица, такие как большой нос и заниженные веки.
3. Очно-зрительные нарушения: многие дети с синдромом Тричера-Коллинза имеют проблемы с зрением, такие как катаракта, глаукома или аномалии роговицы. Они могут иметь также проблемы с слухом и речью.
4. Нервно-психические расстройства: дети с этим синдромом часто страдают от психических расстройств, таких как умственная отсталость, расстройства аутистического спектра, проблемы с остроумием и социальным взаимодействием.
5. Дополнительные физические аномалии: некоторые дети с синдромом Тричера-Коллинза могут иметь другие физические аномалии, такие как пороки сердца, нарушения речи и языка, нарушения нервной системы и суставные проблемы.
У каждого ребенка с синдромом Тричера-Коллинза могут быть разные комбинации симптомов, и их тяжесть может различаться. Все симптомы нужно наблюдать именно в комплексе, так как это набор особенностей, характерных для данного синдрома. Если у вашего ребенка есть подозрение на синдром Тричера-Коллинза, важно обратиться к врачу-генетику для получения точного диагноза и разработки соответствующего плана лечения и ухода.
Влияние на психическое состояние человека
Синдром Тричера-Коллинза является серьезным заболеванием, которое оказывает значительное влияние на психическое состояние человека. Пациенты, страдающие от этого синдрома, испытывают постоянные эмоциональные переживания, беспокойство и тревогу.
Данный синдром сопровождается нарушением функций лобных лжецентров, что приводит к нарушению планирования повседневных дел, контроля над собственными поступками и принятия решений. Пациенты часто испытывают проблемы с концентрацией, запоминанием новой информации и одновременным выполнением нескольких задач.
Такие нарушения обычно вызывают социальную дезадаптацию и серьезные проблемы в повседневной жизни. Пациенты могут испытывать трудности в установлении и поддержании отношений, испытывать затруднения в организации своего времени и управлении своими эмоциями.
Однако, с помощью комплексного лечения и поддержки со стороны специалистов, возможно значительно улучшить психическое состояние пациентов с синдромом Тричера-Коллинза. Лечение может включать в себя применение лекарственных препаратов, психотерапевтические методики, а также специальные упражнения и тренировки для улучшения когнитивных функций.
Причины развития синдрома Тричера-Коллинза
Синдром Тричера-Коллинза, также известный как синдром дополнительной 15 хромосомы или синдром интеллектуальной недостаточности, является генетическим нарушением, которое возникает из-за наличия лишнего фрагмента хромосомы 15. В то время как точные причины появления этого синдрома до конца не изучены, некоторые исследователи указывают на следующие факторы.
- Генетические нарушения: Синдром Тричера-Коллинза обусловлен наличием лишнего фрагмента хромосомы 15. Это может произойти из-за различных генетических нарушений, включая мутации, делеции или транслокации хромосом.
- Семейная предрасположенность: В некоторых случаях синдром Тричера-Коллинза может быть унаследован от родителей, у которых есть генетические нарушения связанные с хромосомой 15.
- Мозаицизм: В некоторых случаях мутация хромосомы 15 может происходить на ранних стадиях развития эмбриона. Это может привести к мозаицизму, когда некоторые клетки имеют нормальное количество хромосом, а другие содержат дополнительный фрагмент хромосомы 15.
- Внутриутробные факторы: Некоторые исследования показывают, что воздействие внутриутробных факторов, таких как инфекции или токсические вещества, может увеличить риск развития синдрома Тричера-Коллинза.
В целом, причины развития синдрома Тричера-Коллинза до сих пор не полностью изучены и требуют дальнейших генетических исследований. Однако, эти факторы могут играть роль в возникновении данного генетического нарушения.
Генетическая предрасположенность к синдрому Тричера-Коллинза
Синдром Тричера-Коллинза является генетическим заболеванием, которое передается по наследству от родителей к ребенку. Наследование этого синдрома происходит по преимущественно аутосомно-доминантному типу. Это означает, что для возникновения синдрома достаточно наличия только одной мутированной копии соответствующего гена.
Причиной развития синдрома Тричера-Коллинза является мутация гена TRPS1, который находится на одной из хромосом. Эта мутация приводит к нарушению нормального функционирования гена и формированию характерных симптомов синдрома. Мутация может возникнуть как спонтанно, так и быть унаследованной от одного из родителей, которые самостоятельно страдают от синдрома.
Исследования показывают, что генетическая предрасположенность к синдрому Тричера-Коллинза наблюдается в семьях, где уже есть случаи этого заболевания. В таких семьях риск появления синдрома у наследников увеличивается. Родители, которые сами являются носителями мутации гена TRPS1, имеют 50% вероятность передать ее своим детям.
Важно отметить, что синдром Тричера-Коллинза является редким заболеванием и встречается среди населения с низкой частотой. Однако, для семей, имеющих генетическую предрасположенность к этому синдрому, рекомендуется проведение генетического консультирования и тестирования перед планированием беременности. Это позволит определить наличие мутации у родителей и оценить риск передачи ее детям.
Внутренние и внешние факторы, способствующие развитию синдрома
Развитие синдрома Тричера-Коллинза обусловлено взаимодействием различных внутренних и внешних факторов. Внутренние факторы, такие как генетическая предрасположенность и нарушения в структуре головного мозга, могут играть роль в возникновении синдрома. У некоторых людей может быть увеличенный размер задней части головного мозга или другие аномалии, которые могут повлиять на развитие синдрома.
Внешние факторы также могут способствовать развитию синдрома Тричера-Коллинза. Ранняя недостаточность питания и витаминной поддержки во время беременности или в раннем детстве может повлиять на развитие мозга и функционирование нервной системы. Травмы головы, инфекции или другие медицинские проблемы также могут быть факторами, способствующими развитию синдрома.
Кроме того, социальная среда и условия жизни могут оказывать влияние на развитие синдрома Тричера-Коллинза. Недостаточное качество медицинской помощи, отсутствие доступа к услугам ранней интервенции и поддержке в обучении могут значительно ограничить возможности развития ребенка и привести к возникновению синдрома.
Диагностика синдрома Тричера-Коллинза
Диагностика синдрома Тричера-Коллинза основывается на физическом обследовании пациента и анализе его медицинской и семейной истории. Врачу важно обратить внимание на характеристики лица ребенка, особенно наличие специфических признаков, таких как низкое лоб, маленький размер нижней челюсти, широко расставленные глаза и неправильные зубы.
Для уточнения диагноза может быть проведена генетическая консультация и генетическое тестирование, которое позволяет выявить наличие мутаций в генах, ответственных за развитие синдрома Тричера-Коллинза. Кроме того, могут использоваться другие инструменты диагностики, такие как рентгенография костей черепа и лица, компьютерная томография головного мозга и сердца, электрокардиография.
Важную роль в диагностике синдрома Тричера-Коллинза играет оценка развития речи и психомоторного развития ребенка, а также проведение неврологического обследования. Эти методы позволяют определить наличие задержки в развитии и выявить другие признаки синдрома, такие как нарушение координации движений, задержка роста и развития и другие.
Профилактика и предотвращение синдрома Тричера-Коллинза
Профилактика синдрома Тричера-Коллинза направлена на предотвращение развития данного заболевания и улучшение качества жизни пациента. Для этого следует соблюдать ряд рекомендаций и проводить профилактические мероприятия.
Одной из основных мер профилактики является регулярное проведение медицинских обследований для своевременного выявления возможных нарушений в работе организма. Важно обратить внимание на состояние нервной системы и обмена веществ, так как именно эти факторы имеют важное значение в развитии синдрома Тричера-Коллинза.
Также следует уделить внимание поддержанию здорового образа жизни, включающего регулярное физическое упражнение, правильное питание и отказ от вредных привычек. Физическая активность помогает укрепить организм, улучшить работу сердца и сосудов, а также поддержать нервную систему в хорошем состоянии.
Значительное влияние на профилактику и предотвращение синдрома Тричера-Коллинза оказывает и психологическое состояние человека. Регулярное посещение психолога или психотерапевта помогает контролировать стрессовые ситуации, развивать стратегии управления эмоциями и снижать уровень тревожности.
Важно также обратить внимание на окружающую обстановку и создать комфортные условия для жизни. Это включает устранение факторов риска, таких как пыль, химические вещества и шум. Также нужно регулярно проводить проветривание и поддерживать оптимальную влажность в помещении.
И, наконец, регулярное применение рекомендованных лекарственных препаратов и соблюдение всех рекомендаций врача также является важным моментом в профилактике и предотвращении синдрома Тричера-Коллинза.
Методы лечения синдрома Тричера-Коллинза

Синдром Тричера-Коллинза является разновидностью расстройства аутистического спектра и требует комплексного лечения. Основная цель терапии — улучшение качества жизни пациента и снижение его симптомов, таких как социальные и коммуникативные проблемы, стереотипное поведение и явные высказывания.
Один из ключевых аспектов лечения синдрома Тричера-Коллинза — это поведенческая терапия, которая базируется на принципах приложения отрицательного и положительного подкрепления. Во время сеансов пациенту помогают осознать и контролировать свои эмоции, а также улучшить навыки социального взаимодействия.
Помимо этого, пациентам с синдромом Тричера-Коллинза может быть назначена лекарственная терапия. Она направлена на смягчение симптомов и улучшение качества жизни. Некоторые препараты могут помочь справиться со структурными или химическими изменениями в мозге, связанными с данным синдромом.
Также важной составляющей лечения синдрома Тричера-Коллинза является речевая терапия. Специалисты помогают пациенту развивать и улучшать коммуникативные навыки, а также научиться использовать язык для выражения своих мыслей и желаний. Регулярные занятия с речевым терапевтом могут значительно улучшить способность пациента к общению.
Одним из дополнительных методов лечения синдрома Тричера-Коллинза является использование техники активационной игровой терапии. Эта методика позволяет пациенту развивать когнитивные и моторные навыки через игры и активные упражнения. Такой подход повышает мотивацию пациента к обучению и способствует лучшему усвоению нового материала.
Видео по теме:
Вопрос-ответ:
Что такое синдром Тричера-Коллинза?
Синдром Тричера-Коллинза — это редкое врожденное заболевание, которое характеризуется отставанием в физическом и умственном развитии ребенка, а также аномалиями внешности.
Какие симптомы характерны для синдрома Тричера-Коллинза?
Симптомы синдрома Тричера-Коллинза могут включать такие проявления, как нарушения в развитии лица (вкл. аномалии глаз, носа, челюстей), задержка в физическом развитии, умственная отсталость, проблемы с речью, эпилепсия и другие.
Последствия нераспознанного и нелеченного синдрома Тричера-Коллинза
Синдром Тричера-Коллинза является серьезным заболеванием, которое оказывает негативное влияние на психическое и физическое развитие человека. Но, кроме этого, нераспознанный и нелеченный синдром может привести к ряду серьезных последствий.
Одной из основных проблем, с которой сталкиваются пациенты с синдромом Тричера-Коллинза, является задержка в развитии речи и коммуникативных навыков. Они испытывают трудности в понимании и использовании языка, что затрудняет их социальное взаимодействие и обучение. Более того, нераспознанный синдром может привести к развитию дополнительных психических расстройств, таких как депрессия и тревожные состояния.
Одним из главных последствий нераспознанного и нелеченного синдрома Тричера-Коллинза является ограничение физической активности и нарушение координации движений. Пациенты испытывают трудности в самостоятельном передвижении и выполнении обычных жизненных задач. Это ограничение мобильности влечет за собой проблемы в самообслуживании, а также приводит к социальной изоляции и понижению самооценки.
Нераспознанный синдром Тричера-Коллинза также может повлечь за собой проблемы со зрением, слухом и здоровьем в целом. Пациенты могут испытывать затруднения в видении и слухе, что затрудняет их обучение и общение с окружающими. Более того, они подвержены различным медицинским проблемам, таким как сердечные заболевания, нарушения пищеварения и проблемы с дыханием.
Очень важно распознать синдром Тричера-Коллинза в раннем возрасте и начать своевременное лечение. Это поможет предотвратить возникновение серьезных последствий и дать пациенту возможность развиваться и достигать своего потенциала по максимуму.
Современные исследования и перспективы лечения синдрома Тричера-Коллинза

Синдром Тричера-Коллинза, также известный как синдром Янца, является генетическим расстройством, которое характеризуется различными физическими и умственными особенностями. В настоящее время проводятся исследования по пониманию причин и механизмов возникновения этого синдрома.
Одна из перспективных областей исследований — генетическая терапия. Ученые изучают гены, связанные с синдромом Тричера-Коллинза, и ищут возможные способы замены или исправления дефектных генов. Это может открыть новые возможности для лечения и предотвращения синдрома в раннем возрасте.
Другим направлением исследований является разработка инновационных методов реабилитации и обучения для людей с синдромом Тричера-Коллинза. С помощью передовых технологий, таких как виртуальная реальность и компьютерные программы, можно улучшить психомоторные навыки и когнитивные функции пациентов, а также способствовать их социальной интеграции.
Однако, несмотря на все перспективы, в настоящее время нет специфического лекарства или метода лечения, направленного на устранение синдрома Тричера-Коллинза. Лечение этого состояния является комплексным и включает в себя поддержание здоровья, реабилитацию и специализированную помощь, а также психологическую поддержку для пациентов и их семей.
Синдром Тричера Коллинза, также известный как челюстно-лицевой дизостоз, является редким генетическим заболеванием, вызванным мутацией в пятой хромосоме
Он никак не влияет на умственное развитие ребенка, характеризуется частичной атрофией скул, деформацией нижней челюсти, раздвоением век и запавшими косыми глазами. Из-за искаженных ушных раковин у пациентов часто возникают проблемы со слухом или полная глухота.
Проблемы людей с синдромом Тричера Коллинза
Помимо очевидных физических недостатков, болезнь приносит эмоциональные проблемы – люди с синдромом Тричера Коллинза часто имеют низкую самооценку и страдают неуверенностью в себе. Их нетипичный внешний вид зачастую вызывает неоднозначное отношение окружающих и может привести к социальной изоляции, что особенно тяжело для детей и подростков. «Люди без лица» обычно не любят смотреть в зеркало и могут чувствовать себя чужими в мире, где нет никого, похожего на них.
О том, как живут люди с синдромом Тричера Коллинза, можно узнать, посмотрев мелодраму Стивена Чбоски «Чудо-мальчик» – экранизацию самого продаваемого романа американской писательницы Р. Дж. Паласио, разошедшегося миллионными тиражами по всему миру. Его главный герой Огги Пуллман – пятиклассник с синдромом Тричера-Коллинза, который не расстается со своим игрушечным шлемом астронавта, позволяющим избегать любопытных, и не всегда доброжелательных, взглядов прохожих. За первые 10 лет жизни мальчик перенес 27 операций, позволивших ему дышать, видеть и слышать.
Содержание
- Причины появления аномалии
- Признаки синдрома Тричера Коллинза
- Диагностика
- Эффективность лечения
- Возможные осложнения и прогноз
- Рекомендации по профилактике
- Отзывы
Синдром Тричера Колинза – врожденная генетическая патология, приводящая к нарушению строения лицевой части черепа. Название заболевание получило в честь врача, впервые подробно описавшего расстройство в 1900 году. Вклад в изучение аномалии внес и другой человек – Франческетти. Этим объясняется вариабельность термина, обозначающего одну и ту же проблему. У пациентов с патологией сразу после рождения диагностируются характерные изменения лица, включающие микрогнатию (маленькую нижнюю челюсть), деформацию скуловых костей и орбит. Зачастую отмечаются нарушения слуха различной интенсивности. Лечение основано на осуществлении хирургической коррекции костных структур.
Причины появления аномалии
Заболевание имеет генетическую природу. Дефекты строения костей возникают на фоне мутации структуры, расположенной на длинном плече 5-ой хромосомы. Повреждается участок ДНК, отвечающий за синтез транспортного белка, – соединения, принимающего активное участие в процессах эмбриогенеза. Подобная причина синдрома Тричера Коллинза обеспечивает отсутствие адекватной терапии, поскольку скорректировать изменения невозможно.
Заболевание наследуется по аутосомно-доминантному типу. При этом до 60% случаев диагностики поражения сопряжено с выявлением спонтанных мутаций, то есть деформации черепа появляются у детей, семейный анамнез которых не осложнен хромосомными аномалиями. Точная причина возникновения нарушений на сегодняшний день неизвестна. Предполагается триггерное воздействие неблагоприятных факторов внешней среды и стресса на будущую мать в период беременности, а также прием сильнодействующих лекарственных средств. Генетический дефект характеризуется высокой пенетрантностью, то есть при наличии аномалии в строении хромосомы происходит формирование симптомов. Интенсивность клинических проявлений во многом зависит от типа мутации. В ряде случаев у пациентов отмечаются лишь незначительные изменения формы лицевых костей. Другие же страдают от потери слуха, офтальмологических заболеваний, а также имеют специфическую внешность. Фото больных с синдромом Франческетти широко распространены в Интернете. На телеканале «ТЛС» шел целый цикл телепередач, в которых освещалась жизнь людей с различными редкими патологиями. На экранах были продемонстрированы и пациенты с челюстно-лицевым дизостозом.
Признаки синдрома Тричера Коллинза
Интенсивность проявления генетического дефекта значительно варьируется. В медицине описаны основные симптомы заболевания, используемые для постановки диагноза:
- Недоразвитость челюстей – микрогнатия. У ребенка формируется очень узкий подбородок и маленький рот. Данные изменения могут сопровождаться нарушениями строения зубного ряда.
- Веки вследствие возникновения хромосомной аномалии приобретают треугольную форму. Такое состояние обозначается термином «коломба».
- Скуловые кости и орбиты глаз также уменьшены в размере. Это придает внешности пациентов специфический вид. Лицо кажется «затонувшим» за счет глубоко посаженных органов зрения и крупного носа.
- Многие дети рождаются с дефектом неба – «волчьей пастью». Данная аномалия приводит к затруднению приема пищи, поскольку происходит формирование канала между ротовой и носовой полостью.
- Значительно страдают органы слуха. Это проявляется тугоухостью, вплоть до полной глухоты. Нарушение связано с изменением нормального строения анализатора. Косточки, обеспечивающие восприятие звуков, деформированы, при этом даже осуществление операции не приводит к восстановлению слуха. У некоторых пациентов диагностируется недоразвитость или полное отсутствие ушных раковин.
- В ряде случаев синдром Тричера Коллинза сопряжен с опасными болезнями, например, пороками развития сердца. Тогда прогноз значительно ухудшается. У некоторых детей при рождении обнаруживается полное заращение носовых ходов (атрезия хоан), сопровождающееся респираторными нарушениями.
Согласно имеющейся статистике, лидирующую позицию по распространенности среди клинических проявлений синдрома Тричера Коллинза занимает недоразвитость или гипоплазия костей скулового комплекса, диагностируемая у 81% пациентов. При этом структуры нижней челюсти деформированы и уменьшены в 78% случаев. Подобные нарушения приводят к развитию таких проблем, как изменение прикуса. У большинства детей с патологией он открытый, что может затруднять прием пищи. 28% пациентов страдают от чрезмерно высокой небной дуги, в которой формируется расщелина. Данная патология носит название «волчья пасть».

Со стороны органов зрения у людей с синдромом Коллинза регистрируется уменьшение наклона пальпебральной щели. Такая аномалия диагностируется в 89% случаев и приводит к развитию офтальмологических симптомов. У 69% пациентов отмечается коломба век – дефект, при котором часть структуры отсутствует.
При рентгенографическом исследовании, а также применении КТ врачи выявляют многочисленные изменения со стороны органов слуха. Дети с синдромом рождаются со сращением молоточка, наковальни и стремечка. В ряде случаев костные структуры могут отсутствовать вообще. Подобные дефекты приводят к тугоухости или полной глухоте.
Диагностика
Заподозрить формирование недуга можно еще на этапе эмбрионального развития. Характерные изменения лицевых костей черепа видны на УЗИ. Для подтверждения наличия генетического заболевания потребуется проведение кариотипирования. Неонатальная диагностика синдрома Франческетти-Коллинза основана на специфических клинических признаках. Важным этапом обследования является оценка работы слухового анализатора. В ряде случаев для исключения других возможных проблем проводится клиническое и биохимическое исследование крови, УЗИ, а также магнитно-резонансная и компьютерная томография. Окончательный диагноз ставится на основании генетических тестов.
Эффективность лечения
Специфических методов борьбы с заболеванием на сегодняшний день не разработано. Это связано с генетической этиологией поражения. Основу лечения синдрома Тричера Коллинза составляют различные хирургические техники, направленные, в первую очередь на устранение аномалий строения лица. Пластика позволяет не только улучшить внешний вид пациента, но и восстановить нормальную проходимость носовых каналов. Операция требуется и при наличии «волчьей пасти». Помощь стоматологов необходима для восстановления нормального прикуса и устранения других дефектов зубного ряда, что значительно улучшает качество жизни пациентов. Как правило, лечение проводится поэтапно, согласно возрасту больного. При тугоухости оправдано слухопротезирование, позволяющее компенсировать деформации. В ряде случаев требуется проведение гастротомии для обеспечения полноценного питания ребенка, а также трахеотомии, необходимой при респираторных нарушениях. Важный этап лечения составляют занятия с логопедом и дефектологом. Они способствуют социализации ребенка. Во многих случаях требуется также общение с психологом.
Медикаментозная терапия при патологии практически не используется, что связано с отсутствием видимых результатов. Исключение составляют лишь анальгетические средства, применяемые в период восстановления после травмирующих операций, а также антибиотики широкого спектра, используемые с целью подавления роста болезнетворной микрофлоры.
Возможные осложнения и прогноз
Исход недуга зависит от степени выраженности патологических изменений. Как правило, костные деформации не несут угрозы жизни и здоровью ребенка. Малыши не отстают в умственном развитии, однако испытывают трудности с социальной адаптацией, как из-за специфической внешности, так и в результате нарушений слуха и речи.
Опасными могут быть сочетающиеся с синдромом Фраческетти пороки сердца, а также дефекты костей, приводящие к затруднению дыхания пациента. В таких случаях прогноз более осторожный. Большинство проблем, возникающих на фоне этой генетической мутации, поддаются хирургическому лечению.
Рекомендации по профилактике
Специфических методов предупреждения развития синдрома Тричера Коллинза не разработано. Эта особенность связана с тем, что контролировать хромосомные мутации не удается. Поэтому профилактика возникновения дефектов направлена на правильное планирование беременности. Оно подразумевает проведение кариотипирования родителей с целью выявления хромосомных аномалий. Однако даже такой подход не дает гарантий отсутствия заболевания у будущего ребенка. Это связано с широкой распространенностью спонтанных мутаций, приводящих к формированию дизостоза.
Отзывы
Борис, 28 лет, г. Псков
Сын родился со специфическим строением лица. Врачи поставили диагноз «синдром Тричера Коллинза». Это генетическое поражение, приводящее к изменению строения черепа. Сыну это жить не мешает, поэтому доктора рекомендовали только наблюдение. Никаких других отклонений у ребенка не нашли. Пластику можно будет сделать, когда он подрастет.
Светлана, 32 года, г. Воронеж
Еще во время беременности врачи заподозрили у малыша генетическое заболевание. У плода было изменено строение лица. После рождения дочки доктора подтвердили синдром Тричера Коллинза. Дефекты выражены не очень сильно, однако девочка плохо слышит, поэтому мы пользуемся слуховым аппаратом. Подумываем о косметических операциях, когда дочка станет постарше.